...on the rationalisation, design, optimisation and synthesis of novel phosphonium derived anti-cancer drugs
 |
Establishing Anti-Cancer Phosphonium Salt Structure-activity RelationshipsPart 5by Phil |
Synthesis
5.1 Pre-clinical Candidate Optimisation
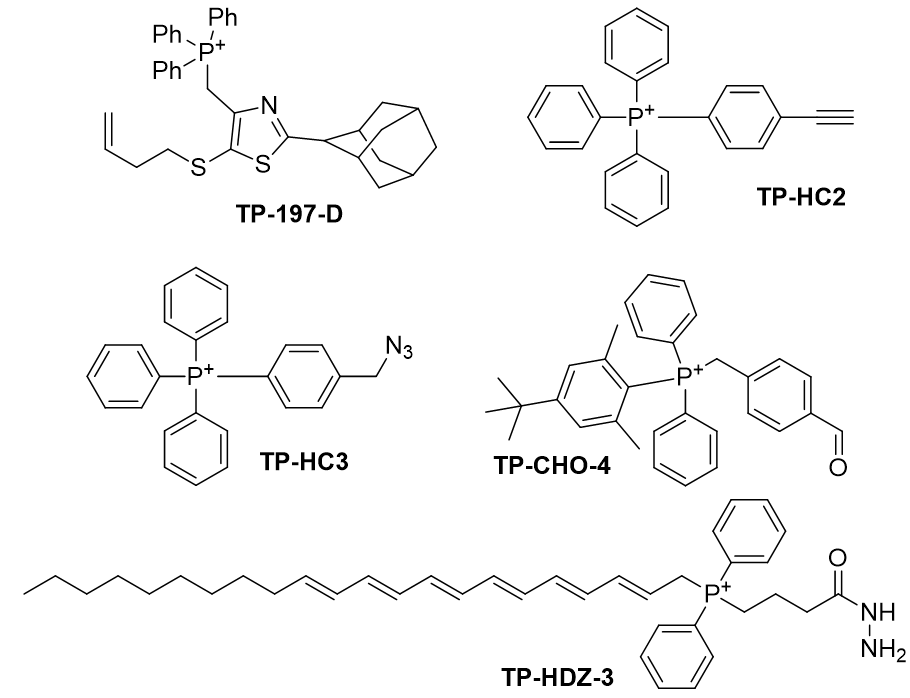
Figure 26 | The five candidates for synthesis
Synthesis of TP-197-D
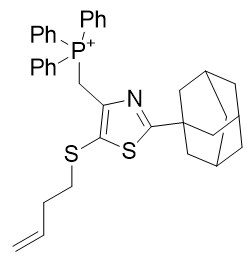
Figure 27 | TP-197-D
Retrosynthesis of TP-197-D reveals a direct route from a thioamide, constructing the thiazole ring and then modifying to meet our substitution requirements.
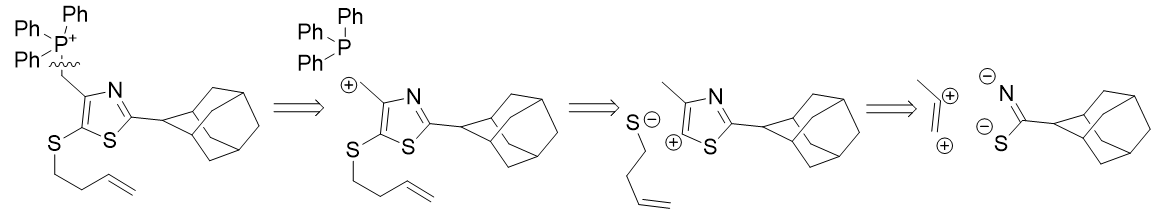
Scheme 3 | Retrosynthetic analysis of TP-197-D
The synthons correlate to available synthetic equivalents (Figure 28).
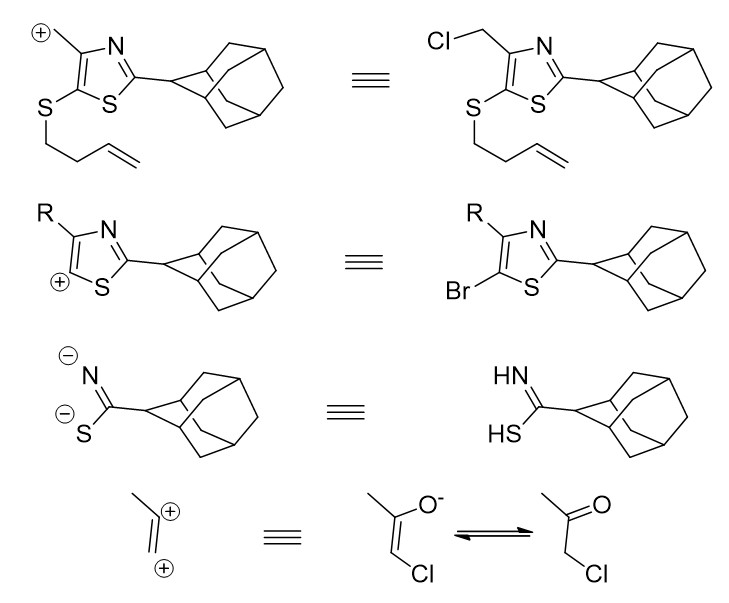
Figure 28 | Synthetic equivalents of the revealed synthons
A forward synthesis can now be proposed. Both aromatic and aliphatic thioamides can be prepared from carbonyl compounds using routes including Lawesson’s sulfurization (route B), Willgerodt-Kindler oxidation (route A) and thiolysis (route C). [68]
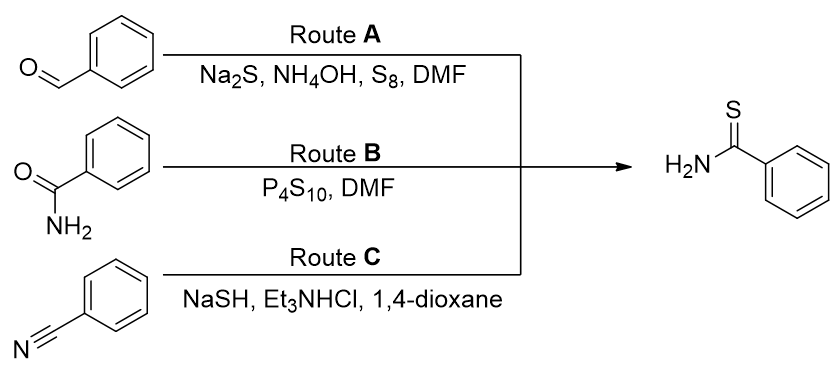
Scheme 4 | Thiamide formation
Reaction with α-haloketones affords thiazoles. [69] The use of bis(cloromethyl) ketone also installs the phosphonium linkage point. Alternative routes include those via 5-carboxythioxazoles esters (not shown). [70]
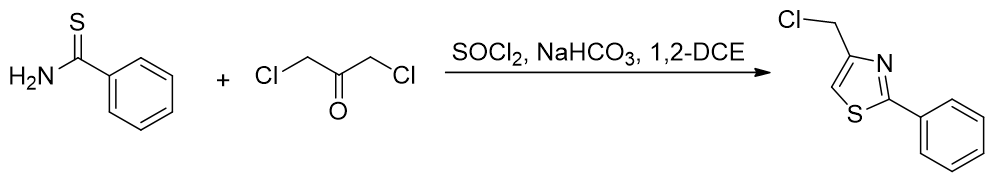
Scheme 5 | Creation of the thiazole ring
Installation of the butylthio group is achieved by non-radical bromination (selective for the thioazole 4-position), [71,72] followed by aromatic nucleophilic substitution. The nucleophilic thiolate is generated from a haloalkane and thiourea. [73]
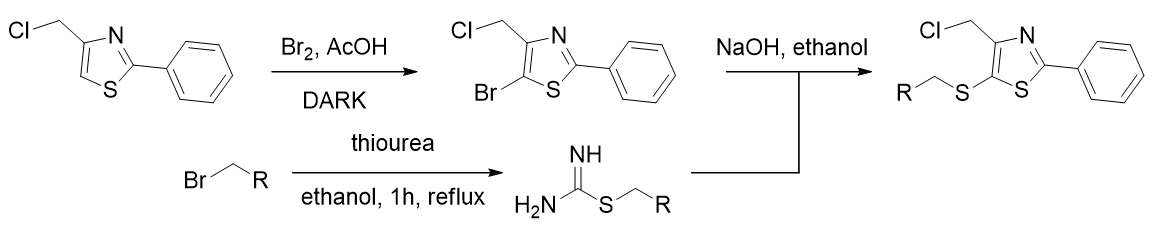
Scheme 6 | Selective ring bromination and incorporation of alkylthiolate
Lastly the phosphonium group is introduced by SN2 Finkelstein reaction, driven the precipitation of LiCl and the intermediate iodide enhanced electrophilicity. [74]
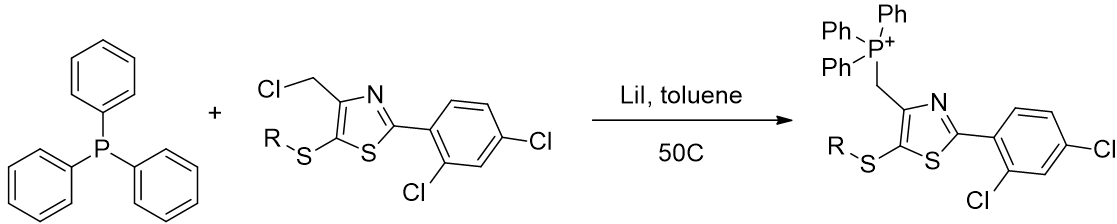
Scheme 7 | Incorporation of triphenylphosphonium
Synthesis of TP-HC2 and TP-HC3
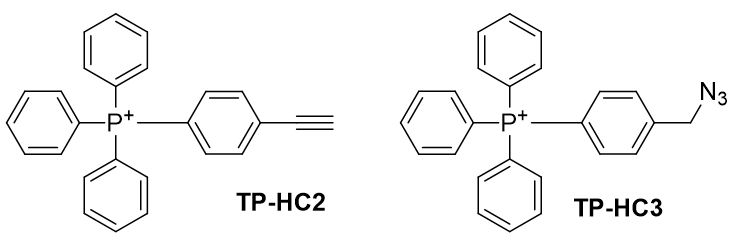
Figure 29 | TP-HC2 and TP-HC3
Simple triphenylphosphoniums are derived from electrophilic aryl species and triphenylphosphine. A source of ‘aryl cations’ are metal mediated coupling reactions.
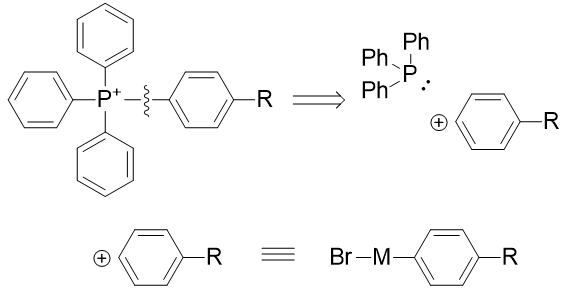
Scheme 8 | Retrosynthetic analysis of simple triphenylphosphonium species
In application to triphenylphosphine, tris(bibenzylideneacetone)dipalladium [75] and niobium bromide [76] have been demonstrated effective. The R group for TP-HC2 is the alkyne and for TP-HC3 is the methylazide.
TP-HC2
Alkynes may not survive the coupling conditions and so is introduced after phosphonium formation by coupling triphenylphosphine with an appropriately functionalised arene. [77]
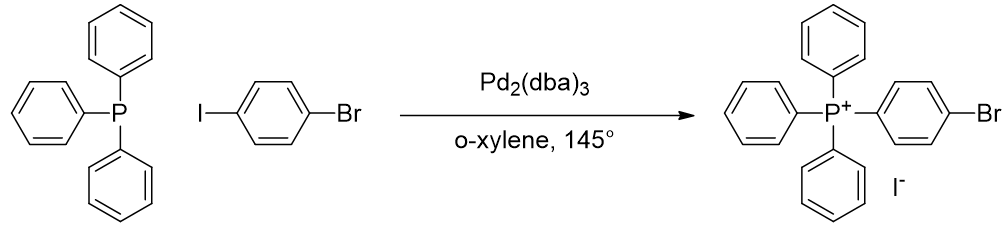
Scheme 9 | p-iodobromobenzene reacts first at the iodine site
Alkynes undergo metal-mediated coupling reactions but coupling of ethyne with copper(i) would be difficult since ethyne is a gas. Trimethylsilylethyne is a liquid equivalent of ethyne but reacts appropriately. [78]
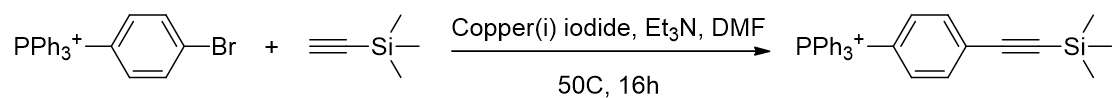
Scheme 10 | Aromatic halide coupling with an ethyne equivalent
The TMS group is removed using potassium carbonate. [79]
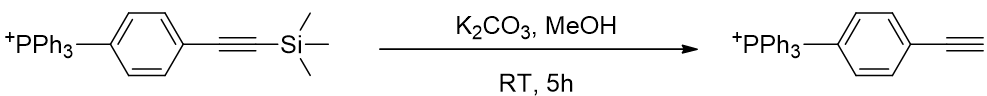
Scheme 11 | TMS deprotection affords TP-HC2
TP-HC-3
Incorporation of the methyl azide is achieved after coupling of the phenyl onto the phosphonium. The bromobenzyl alcohol is coupled using niobium. [80]
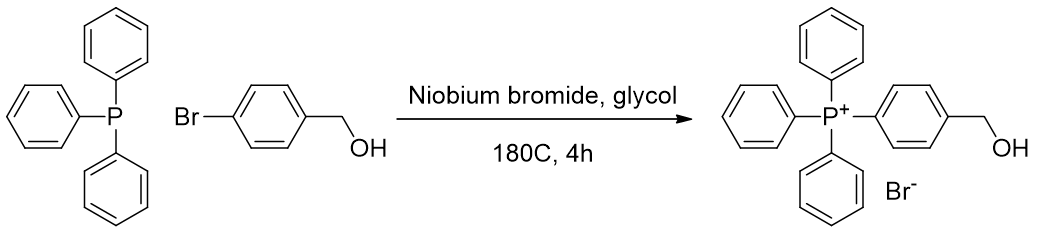
Scheme 12 | Niobium catalysed cross coupling
The alcohol is converted to a chloride with thionyl chloride and then substituted using sodium azide.
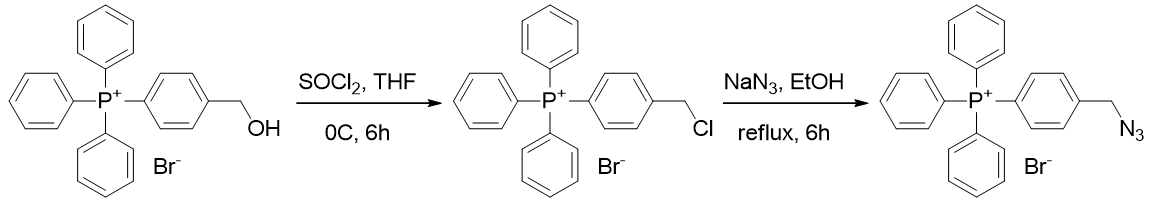
Scheme 13 | Action of thionyl chloride and sodium azide affords TP-HC-3
Synthesis of TP-CHO-4 and TP-HDZ-3
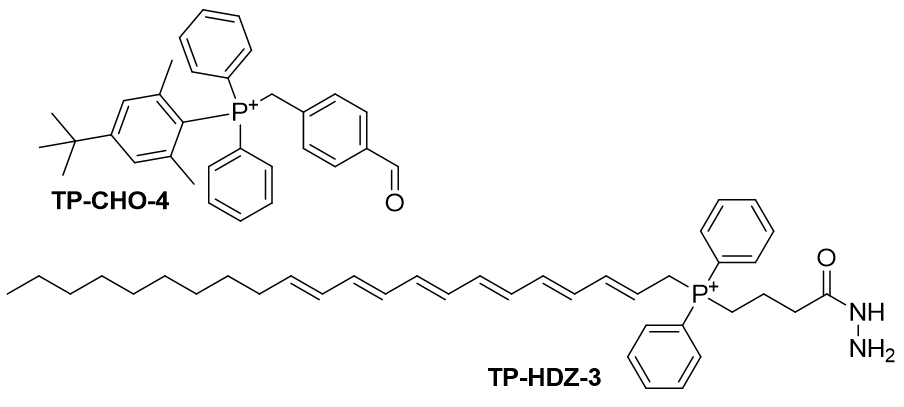
Figure 30 | TP-CHO-4 and TP-HDZ-3
Starting with the electrophilic diphenylphosphorylchloride, [81] reaction with a nucleophilic Grignard reagent affords triarylphosphine.
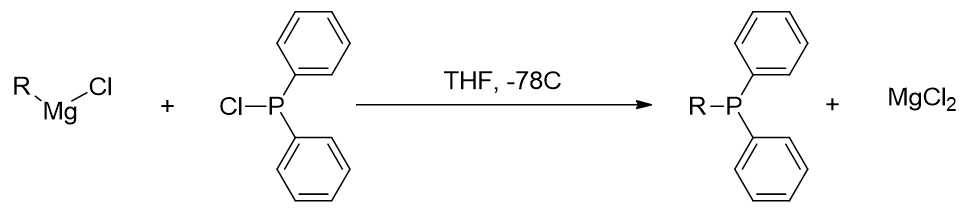
Figure 31 | Reaction of a Grignard Reagent with diphenylphosphorylchloride
The Grignard reagents must first be synthesised using the dissolving magnesium procedure.
TP-CHO-4
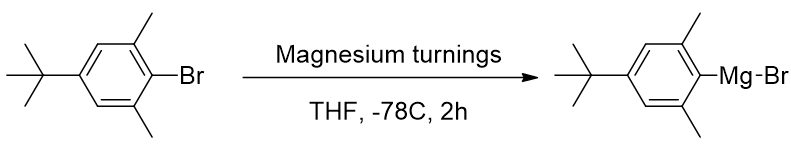
Scheme 14 | Preparation of Grignard reagent for TP-CHO-4
TP-HDZ-3
The lipophilicity of HDZ-3 is derived from the docosahexaenoic acid. Reduction of the acid to the alcohol is achieved using Lithium Aluminium hydride, which is selective over the olefinic positions. Maintaining anhydrous conditions the acid workup is accomplished using ‘acidified ethyl acetate (a mixture of acetic anhydride and ethanol) which avoids π-isomerisation. The alcohol is converted to chloroalkane using thionyl chloride and the Grignard formed using Magnesium.
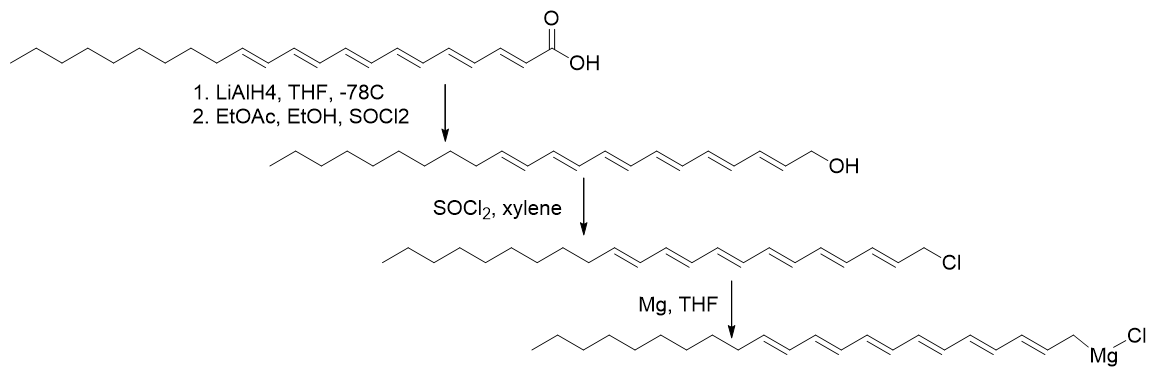
Scheme 15 | Preparation of the Grignard reagent of docosahexaenyl chloride
Reaction of the Grignard reagents with diphenylphosphoryl chloride affords the phosphine starting materials.
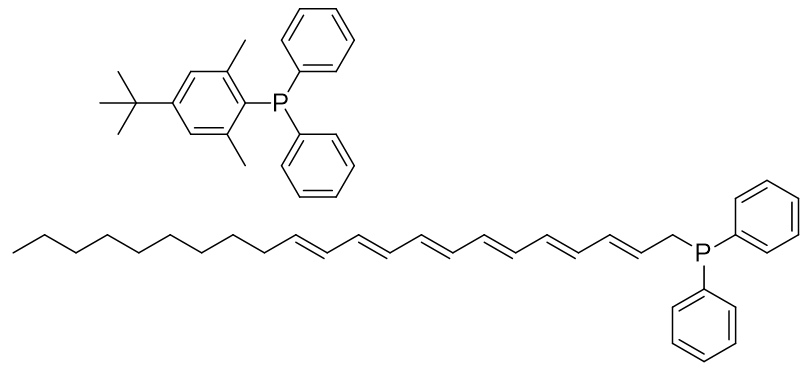
Figure 32 | Phosphine precursors of TP-CHO-4 and TP-HDZ-3
Quaternary substituent incorporation is then accomplished by SN2 reaction. [82]
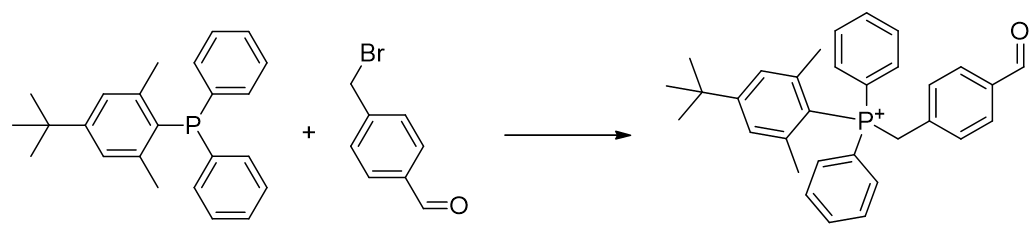
Scheme 16 | SN2 reaction of phosphine affording TP-CHO-4
A direct approach is not appropriate for the hydrazide as the terminal nitrogen is nucleophilic and so must be protected. The t-butylcarbamate group prevents the reactivity of the hydrazide and can be removed by acid once incorporated into the phosphonium.
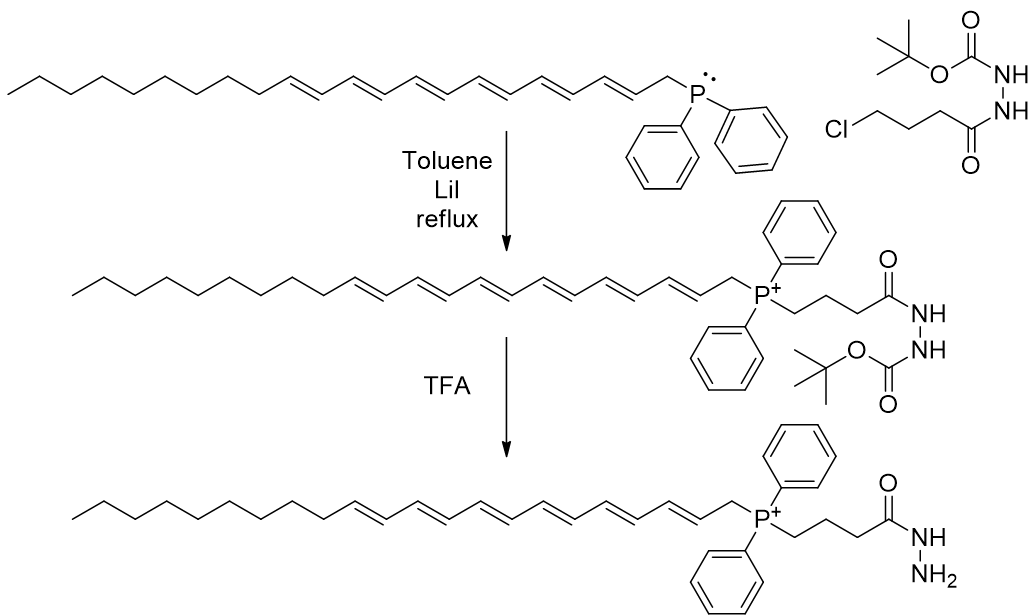
Scheme 17 | SN2 reaction of phosphine to yield TP-HZD-3
5.2 Summary of Synthesis
Several new procedures to phosphonium salts have been proposed. Those of TP-CHO-4 and TP-HDZ-3 appear to be quite universal, modular via various Grignard reagents. ‘Click chemistry’ applied to TPP agents has not been reported, as a first of its kind this is particularly exciting. The compounds TP-HDZ-3 and TD-CHO-4 would make an interesting comparison to those reported in literature, testing the CLogP hypothesis. Finally the heavily modified TP-197-D employs all of the guidelines established.