The current state of melatonin receptor exploration and exploitation: recent and topical advances
 |
The Design of Melatonergic AgonistsPart 1Backgroundby Phil |
This work is dedicated to my grandfather John, who passed away on the 10th of October 2010 after a long struggle with ill-health. Indeed life is a battle we always lose but we shall be damned if we do not go down fighting.
Foreword
Melatonin mediates numerous physiological functions as an agonist at the melatonergic receptors. Discovered nearly half century ago, it is surprising that the pertinence of its action to medicine has only recently been exploited in a designed and systematic fashion.
Unfortunately drug-receptor complexes have proven difficult to obtain in analytic quality and reliable X-ray crystallographic data remains elusive. [38] This has no doubt hindered a more aggressive approach to research the likes of which we've witnessed concerning serotonin. Lacking direct mapping of the human MT receptors, primary research instead relies upon lateral and indirect methods. Particularly enlightening avenues include the establishment of molecular fragment [26,29] and ligand-derivative structure-activity relationships, [5-17] receptor mutagenesis studies and gene sequencing techniques.
Many theories have been formulated on the mode of ligand binding and receptor topology, but as yet there is no universal consensus in the literature. Certain features are now generally agreed upon but there many non-corroborative postulations. It is as the working hypothesis develops that we are able to design more specific and potent drugs. Certainly research is now very active and has expanded rapidly in the recent decades.
The state of the art is such that drug-like activity can be rationalised, potency, kinetics and issues of selectivity at least in part addressed. The efforts of academics and industry have culminated in two new melatonergic agents hitting the market: Ramelteon [27] and Agomelatine. Ramelteon is a chronobiotic agent which advances sleep onset, improves quality of sleep and is novel in that it is neither a sedative nor tranquiliser and lacks their degree of potential for abuse. Agomelatine in addition to its somnial indications, is also the first of a kind in that it is a potent but non-monoaminergic anti-depressant.
 |
 |
 |
| Melatonin | Ramelteon | Agomelatine |
Figure 1 | Melatonergic agonists
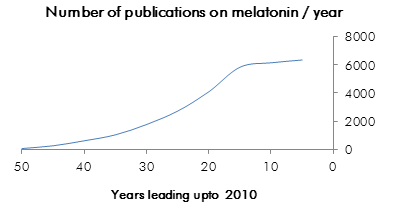
These drugs have already exceeded our early expectations and as new physiological roles of the melatonergic system continue to be discovered, the field is young and prospective gains are plentiful. This is a review of research in the field of melatonergic agonist design, ligand-receptor interaction, receptor topology and agent dynamics.
Contents
Introduction
Melatonin is a neurohormone, principally although not exclusively associated with the pineal gland.[2] Attributed to melatonin are several physiological roles including circadian entrainment and sleep induction. It is both a potent cellular and mitochondrial anti-oxidant, neuro, cardio and hepatoprotective agent, it is an anti-depressant, it is oncostatic and neurotrophic and exerts an effect on the immune system. Indeed melatonin mediates many physiological systems. It is prevalent throughout much of the animal kingdom and abnormal plasma melatonin levels in man have been linked to a variety of disorders. Historically underexploited, the melatonergic system now offers medicine unique opportunities as a vector for novel pharmacotherapeutic intervention.
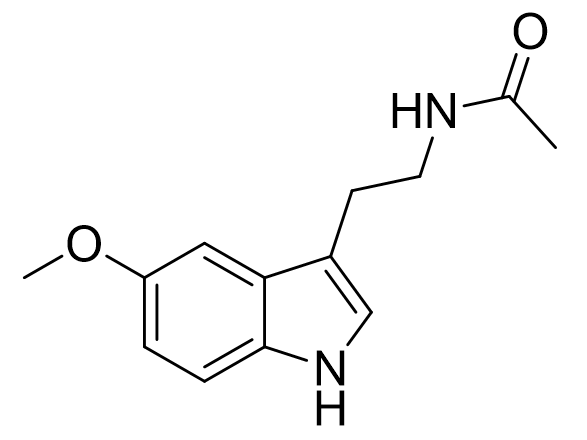
Figure 2 | Melatonin
Dyssomniac conditions often accompany major depressive disorders (MDD), typically as insomnia but also hypersomnia and circadian abnormalities. Profound sleep disturbances in sleep architecture and quality are incident in MDD and other affective disorders. Melatonergic agonists are therefore particularly suitable for treatment of depression with co-morbid sleep dysfunction. Indeed exogenous slow-release melatonin has been employed for this purpose, both useful as a hypnotic and as an anti-depressant.
Background
Higher plasma concentrations are observed during prolonged darkness and thus typically at night-time. This photo-neuroendocrine link is surprisingly not primarily enabled by the image forming cones and rods of the eye, but instead by specialised photosensitive neurones of the retina. These innervate and modulate pineal activity and light exposure acutely suppresses melatonin release. The system is pivotal in the coordination of temporal cycles that can be correlated to changes in luminosity.
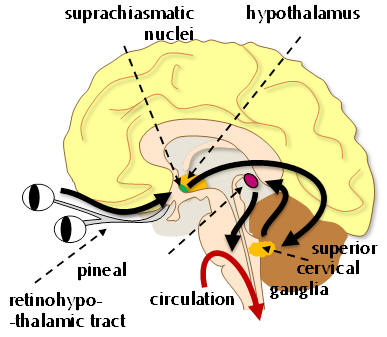 |
Figure 3 | Photo-neuroendocrine transduction: the endocrine function is reflected by the fact that the pineal gland, many of the melatonergic neurones and much of the melatonergic apparatus is localised to the hippocampus, specifically in and around the suprachiasmatic nucleus - a key structure responsible for homeostatic control mechanisms. |
Melatonin has a circadian entraining and sleep inductive effects in man. The use of melatonin has been met with success in the management of age-related onset insomnia and phase-shifted sleep disorders including jet-lag and shift-work. The adjunction of melatonin for the treatment of dysomnia has been commonplace since its role in sleep was establish.
Activity at the MT receptors is responsible for the acute physiological and biochemical effects of melatonergic agonism. The endocrine and neurotransmitter functions of melatonin are specifically enabled by action at the MT1 and MT2 receptors. These are membrane bound G-protein coupled receptors but third melatonin receptor MT3 has now been identified as a soluble quinone reductase.
MT Receptors
In the human physiology, the activity of melatonin is derived from its actions at any of the two distinct but typical melatonin receptors. The distinction between these receptors can be made in terms of ligand binding affinity using Luzindole (N-acetyl-2-benzyltryptamine) and 4P-PDOT (4-phenyl-2-propionamidotetralin). Between these receptors, the constitutional nature differ significantly.[1] These are encoded by the genes Mel1a and Mel1b which correspond to the ML1 and ML2 receptors respectively.
 |
 |
| Luzindole | 4P-PDOT |
Figure 4 | Melatoneric investigatory molecules
Cells of the pituitary gland express the G-protein coupled receptors MT1 and MT2 melatonin receptors. Their activation results in a decrease in cAMP production and a decrease in protein kinase-A activity.[1] Also GnRH-induced gonadotropic secretion is attenuated by melatonin by inhibitory action at both voltage-gated calcium channels and also by prevention of the mobilisation of vescicular calcium.

Figure 5 | MT receptor diagram: the cAMP inhibiting G-protein coupled receptor
Activation of different subtypes of the MT receptors induce different physiological responses. Activation of the MT1 receptors inhibits neuronal activity in the SCN whereas activation of MT2 has been suggested to mediate circadian effects and is involved in dopamine release modulation.
Receptor sub-type selectivity
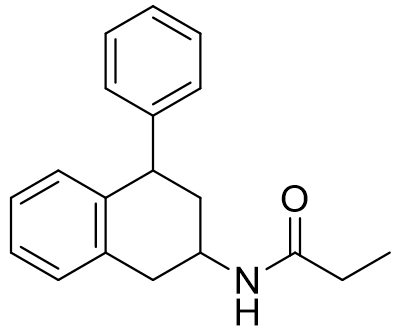
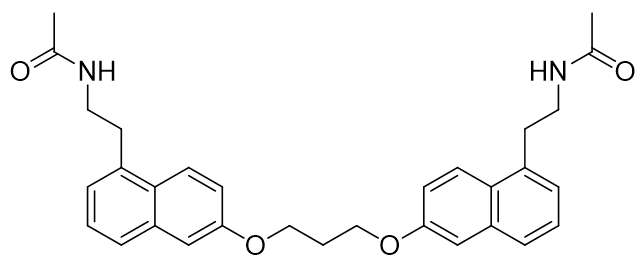
It has been recently proposed that steric factors predominantly determine selectivity for the MT1 receptor. Hence more sterically demanding ligands are generally more selective. This is illustrated with homo-dimeric melatonergic agonists.
 |
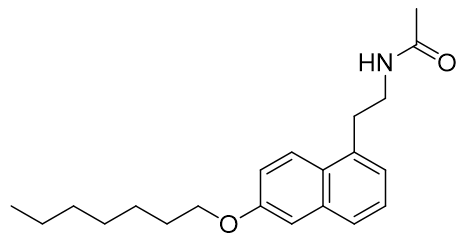 |
Figure 6 | Novel sterically demanding MT1 selective agonists
However without fine detail of either receptor structure available trying to unravel the distinguishing features of each would be rather speculative and so focus will directed towards potency, affinity and kinetics rather than selectivity.
Previous: Summary Presentation | Next: Part 2