The current state of melatonin receptor exploration and exploitation: recent
and topical advances
 |
The Design of Melatonergic Agonists
by Phil
|
New perspectives and novel opportunities for the treatment of sleep
dysfunction and refractory and major depressive disorders
The page contains a summary presentation of the four part essay series.
Over the past 30 years the pharmaceutical industry has been witness
to an explosion of research into the function of melatonin in man and
the use of melatonergic drugs as agents of pharmacotherapeutic and
chronobiotic intervention. Through a series of quantitative structure
activity relationships our understanding of the melatonergic
pharmacophore has been refined. Independent lead optimisation has
culminated in the successful trial and release of two novel melatonergic
drugs, Ramelteon and Agomelatine. Despite conception as a circadian
entraining chronobiotic, the latter has been proven to function beyond
expectation and is hailed as the first non-monoaminergic anti-depressant
and has far reaching implications for medicine of the 21st century.
Whereas melatonin has unfavourable pharmacokinetics, both
Agomelatine and Ramelteon have longer half-lives and are less
disposed to cause nocturnal break-through wakefulness. These
drugs are more lipophilic than melatonin and extensively
distribute into body tissue. Indeed both drugs are more potent
and bind with higher affinities than melatonin.
 |
|
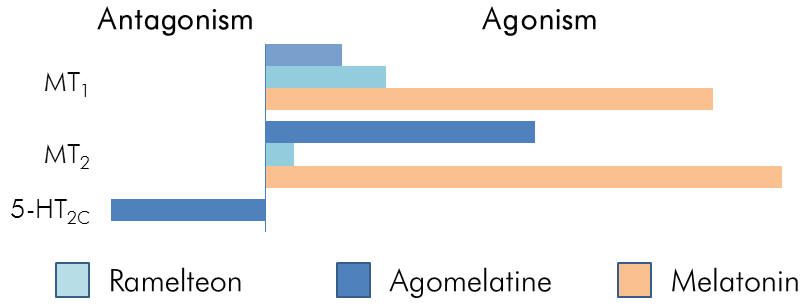
Figure | Comparative pharmacology. EC50 values
determined from radio-ligand binding assays |
 |
|
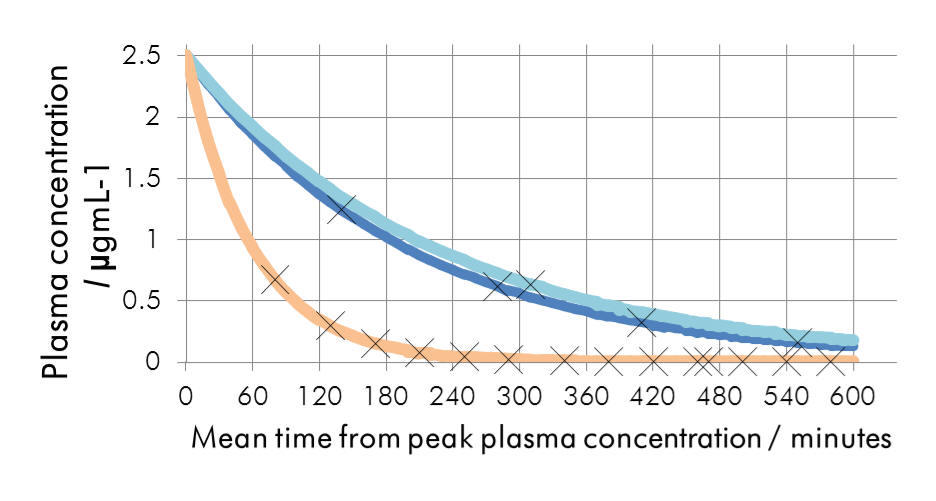
Figure | The elimination kinetics from peak plasma
concentrations of common absolute bio-availabilities |
Having
been proven in the clinical setting, Ramelteon is available as a
chronobiotic and hypnotic in the EU. Agomelatine has also been
found to possess additional antidepressant qualities. Both are
potent MT1/MT2 agonists, but Agomelatine is also a selective
5-HT2C antagonist. This is likely to contribute towards its
anti-depressant efficacy and makes it amenable as an adjunctive
agent for use with SSRIs, potentially offsetting SSRI induced
anxiety.
|
Table 1 | Selected Pharmacological Data /nM |
|
|
T½
|
MT1
IC50
|
MT1
Ki
|
MT2
IC50
|
MT2
Ki
|
Selectivity
|
5-HT2C
IC50
|
5-HT2C
Ki
|
|
Agomelatine
|
2.3h
|
0.13
|
0.10±0.01
|
0.4
|
0.12±0.02
|
3.6
|
270
|
710
|
|
Ramelteon
|
2.6h
|
0.21
|
0.02±0.01
|
0.05
|
0.11±0.05
|
8
|
-
|
-
|
|
Melatonin
|
0.7h
|
0.78
|
0.08±0.02
|
0.90
|
0.38±0.05
|
1.2
|
-
|
-
|
|
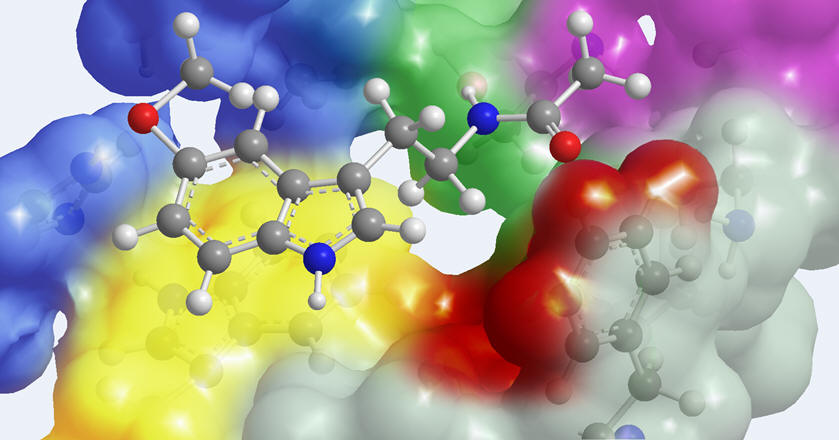
| As reliable X-ray data on human melatonin
receptors remains elusive, much of our understanding of the
essential pharmacophore has been deduced from the extensive
assays of synthetic melatonin derivatives. The goals of such
excursions are four-fold: to understand receptor selectivity, to
design more potent congeners, to predict agonist or antagonist
dynamics & to enhance drug-like pharmacokinetics. |
 |
| |
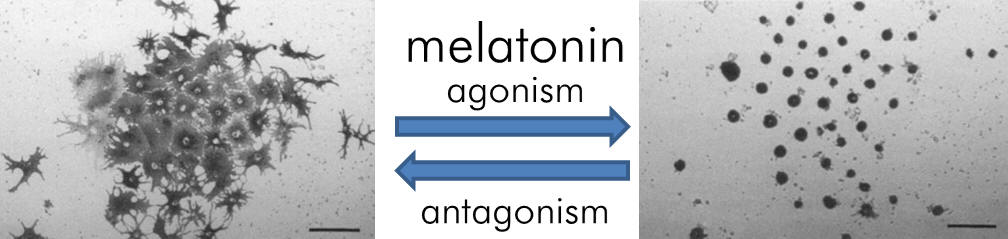
In amphibians melatonin is responsible for night pallor.
This enables the convenient assay of melatonergic activity and
studies on pigment aggregation of xenopus laevis have been
instrumental in developing our understanding.
Sugden et. al., Eur. J. Pharmacol.,
1992, 212(3), 405-408
|
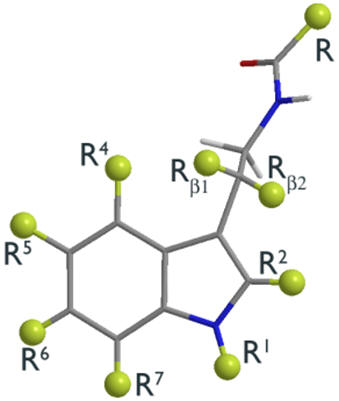
| Melatonin as a structural platform offers many opportunities for
pharmacophore variation. Catenae and annuli homologation, bio-isosteric
substitution, derivatisation and the configuration of absolute
conformation are all tailorable facets. For a more detailed analysis of
the various modifications, see part 3. |
 |
 |
 |
| Peripheral substituents |
Annular atoms |
Di-hedral angles |
 |
|
|
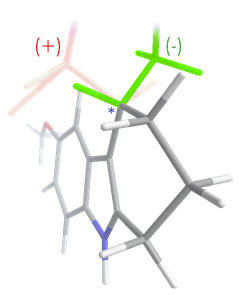 By
restraining the C3 side chain
it has been deduced that
the anti/gauche conformation, with Cβ configured S(-)
enables
optimal location through the amide binding region. By
restraining the C3 side chain
it has been deduced that
the anti/gauche conformation, with Cβ configured S(-)
enables
optimal location through the amide binding region. |
|
 Assay of the two
oxolano- derivatives revealed that in the
active conformation the alkoxy substituent is oriented syn
relative to the C3 chain. This was achieved through cyclisation
of the aliphatic ether via a methylene spacer back onto the
tryptamine e-face. Assay of the two
oxolano- derivatives revealed that in the
active conformation the alkoxy substituent is oriented syn
relative to the C3 chain. This was achieved through cyclisation
of the aliphatic ether via a methylene spacer back onto the
tryptamine e-face. |
|
|
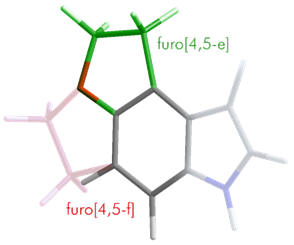
Quite diverse bioisosteres of the pyrrole moiety are
tolerated. Assay of N-capped, substituted and rearranged
derivatives have shown this part of the cyclic system to not be
significantly involved in the binding mode. This gives
opportunities to tailor the pharmacokinetic profile.
| Pyrrole bioisosteres |
 |
 |
 |
 |
 |
| Furyl |
Naphthyl |
Chain transfer |
Cycloalkyl |
N-methyl |
| * all
modifications tolerated, with only mild reductions in
activity |
|
We have come up with two new, potential melatonergic agonists,
which exploit several of the various optimisations.
|
Takeda Pharmaceuticals and Servier Laboratories
arrived at the two effective and novel melatonergic agonists,
Ramelteon and Agomelatine. Ramelteon has the methoxy and amide
side chains constrained into their active conformations whilst
the acetyl group is homologated and both drugs exploit the
tolerability of pyrrole bioisosterism to enhance
pharmacokinetics. As would be expected by homologation and
bioisosteric substitution with lipophilic moieties, both drugs
indeed possess superior pharmacokinetic profiles. Optimisation
of the pharmacophore has produced very potent drugs.
 |
 |
| Ramelteon |
Agomelatine |
 |
|
From our working hypothesis new melatonergic agonists and to
a lesser extent antagonists can be predicted.. Several SARs
studies support the rational design of melatonin agonists.
Ramelteon is a grand example of the success of SARs studies, and
its pharmacokinetics and dynamics can be easily rationalised
relative to that of melatonin. I do wonder why we need a drug to
do what melatonin does rather well; issues of pharmacokinetics
can be addressed through delivery or the optimisation of drug
formulation. Agomelatine is far more structurally simple than
Ramelteon, and it may appear less thought went into its design -
one simple bioisosteric substitution. Not a huge difference
between it and melatonin, but the substitution of NH for CHCH
makes a big impact on patent applications. Indeed Ramelteon may
be a victim of its own ingenuity in that designed as a selective
agent it lacks the broader serotonergic activity for which
Agomelatine is praised and perhaps most famous.
- Agomelatine and Ramelteon pharmacodynamics are
easily rationalised by interpolation with QSAR studies
- These
new drugs are selective melatonergic agonists and may be useful
for the treatment of a range of mental and sleep disorders
- We
can expect a rapid clinical embrace of these new agents but also
an expansion of the array of melatonergic drugs as new congeners
are synthesised
|
Next: Part 1
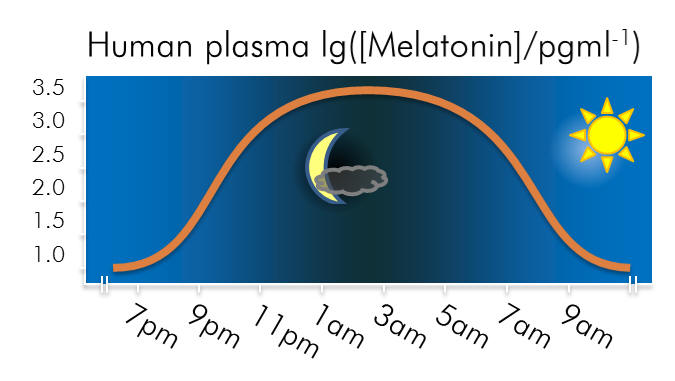
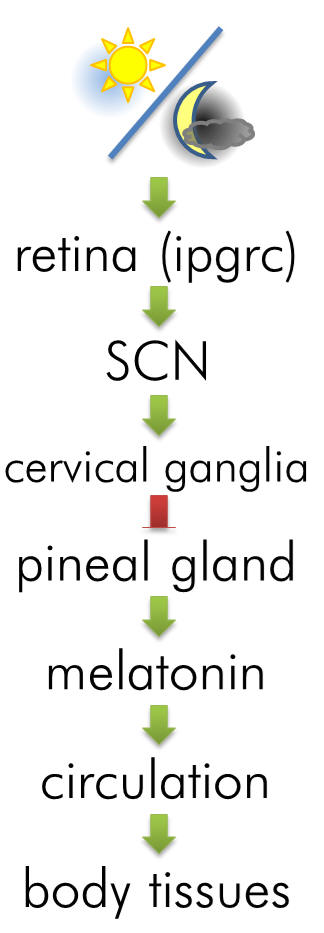
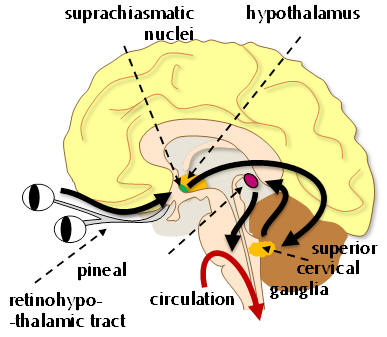
 Melatonin is a neurohormone principally associated with the
pineal gland. As a modulator of the circadian and seasonal
rhythms, an anti-oxidant and antidepressant, melatonin is
extensively exploited physiologically - it is also vasoactive,
oncostatic, cell-protective and neurotrophic. Abnormal plasma
melatonin levels in humans have been linked to many disorders
including phase-shifted sleep disorders, dysomnias and
depressive and cyclic psychological states. Insomnia is often
co-morbid with depression and so the activity of melatonin
offers a unique vector for opportunities of chronobiotic and
pharmacotherapeutic intervention.
Melatonin is a neurohormone principally associated with the
pineal gland. As a modulator of the circadian and seasonal
rhythms, an anti-oxidant and antidepressant, melatonin is
extensively exploited physiologically - it is also vasoactive,
oncostatic, cell-protective and neurotrophic. Abnormal plasma
melatonin levels in humans have been linked to many disorders
including phase-shifted sleep disorders, dysomnias and
depressive and cyclic psychological states. Insomnia is often
co-morbid with depression and so the activity of melatonin
offers a unique vector for opportunities of chronobiotic and
pharmacotherapeutic intervention.
 The latter of these is less well
understood but the former are pertinent to the endocrine and
central nervous system. These are typical cAMP inhibiting 7-TM
G-protein coupled receptors. MT1 agonism activates protein
kinase-B and is responsible for an acute neuronal inhibitory
effect. MT2 agonism induces both soluble guanylate cyclase and
protein kinase-C and is likely to be responsible for circadian
entrainment.
The latter of these is less well
understood but the former are pertinent to the endocrine and
central nervous system. These are typical cAMP inhibiting 7-TM
G-protein coupled receptors. MT1 agonism activates protein
kinase-B and is responsible for an acute neuronal inhibitory
effect. MT2 agonism induces both soluble guanylate cyclase and
protein kinase-C and is likely to be responsible for circadian
entrainment.