The current state of melatonin receptor exploration and exploitation: recent and topical advances
 |
The Design of Melatonergic AgonistsPart 3Derivative investigation studiesby Phil |
Contents
- The rationale for design: why do we need agonists?
- Motivation of SARS studies
- N1-X
- C3
- Acetyl group
- C5-X substitutions
- C5-OR conformation
- Fragment based studies
- C2-X derivatives
- 2-phenyl melatonin sub-series
- Face-A fused polycyclic 2-phenylmelatonin derivatives
- Aliphatic derivatisation
- Fused face-B tricyclic derivatives
- Face-C,D perifused tricyclic derivatives
The rationale for design: why do we need agonists?
Recent meta-analysis has revealed that melatonin is not sufficiently effective in treating primary sleep disorders. This may be due to melatonins very short half-life. To this address, new agents are designed to have appreciable longer half-lives.
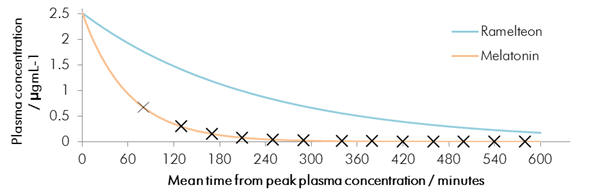
Chart 1 | Melatonin elimination from plasma: t½ ≈ 60min compared to 180min (Ramelteon)
Melatonin does not exhibit good drug like pharmacokinetics nor is it receptor specific and this may cause undesirable side-effects. Melatonin is frequently ascribed a minimal side-effect profile and is considered by many to be very safe. However at large super-therapeutic dosages (1000 fold excess) in some mammals induction of reproductive regression has been observed - this and prenatal complications may be potential side-effects in humans. This may be an avertable consequence when selectivity and receptor discrimination is fully understood.
Medicinal chemists can address these issues and these have acted as the motivation for the design of novel melatonergic agonists.
Motivation of SARS studies
Whilst there is likely to be a significant degree of receptor flexibility there will exist a ligand binding-mode. This would involve the ligand in its 'optimal active conformation', one which is complementary of the receptor topology. This information is typically ascertained from X-ray crystallographic analysis of the receptor-drug complexes, but these complexes in man are difficult to obtain. When no reliable observations are available, a theoretical framework must formulated from what indirect empirical evidence is available.
To identify the features, geometries and configurations which constitute the optimal pharmacophore, a variety of conformationally restrained and substituted ligand derivatives have been reviewed. The objective is to produce a model which fits what evidence is available such that agent dynamics and kinetics can tailored, including understanding and implementing factors affecting adsorption, distribution, elimination, potency, affinity and selectivity.
N1-X and Furan isosteres and substitutions
When alkylated, the potential for H-bonding is obviated and it is surprising that derivatisation of N1 neither prevents binding nor eliminates activity.[25] Diverse pyrrole component variations are tolerated and modest changes do not dramatically affect the drug-receptor binding profile.
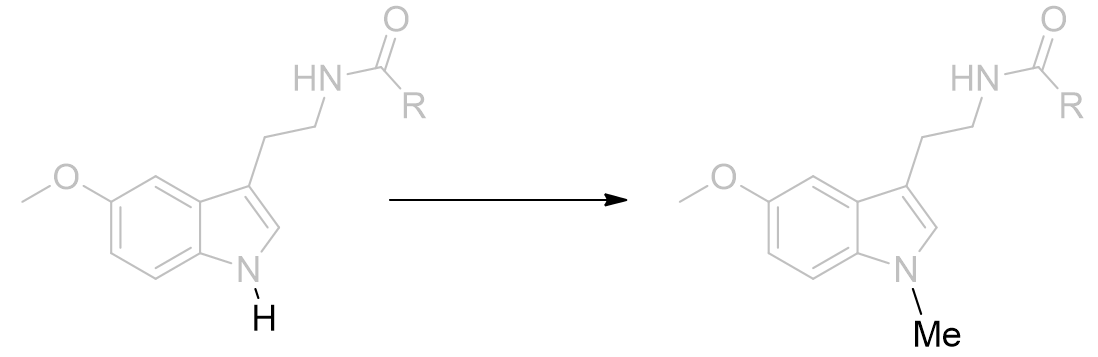
Figure 18 | Melatonin N-methylation: a 40% decrease in agonist dynamic activity results [34]
The binding affinity not dropping by a large amount suggests that H-bonding is unlikely to operate on the pyrrole group, although a less significant role cannot be ruled out. Further testing of this hypothesis is achieved by heteroatom substitutions which exhibit alternative or no H-bonding capacity. Perhaps rather than a formal H-bonding regimen, instead the pocket is hydrophilic and coulombically stabilising. The pocket may be deep or continuous with the C2 environment.
The furan derivative (lacking a polarised hydrogen atom) is not capable of acting as an H-bond donor but still retains good binding capacity. The binding site can accommodate many congeners including ring expansion homologs and those derived through bioisosteric substitutions and aromatic saturation. These results suggest that the receptor response is not directly dependent upon molecular dynamics arising from interaction at this site. Indeed it appears the N1 atom is not required for activity and is not essential for binding. This is quite fortuitous from the perspective of tailoring the pharmacokinetic profile. The pyrrole heterocycle is hydrophilic and this is to the detriment of the drug adsorption and elimination. Thus to the address of melatonins short half-life, this pyrrole cycle can be derivatised, bioisosterically substituted or saturated to result in a more preferable lipophilicity partition co-efficient - the corollary being an improved ADME profile.
 |
 |
 |
 |
| H-bond donor | H-bond acceptor | Non H-bonding | |
Figure 19 | Pyrrolic heteroatom substitutions: the mode of putative H-bonding changes on N1 substitution and so downplays the contribution of pyrrole nitrogen coordination
The last of this series is particularly interesting because with a saturated C3 position this molecule is asymmetric. Thus it offers another variable for the control of the pharmacophore and enables the absolute positioning of the amide chain without the need for an additional constraining bulky fused cycle (vide infra). Whilst no mention of a saturated indole or oxa
C3
The C3 atom is planar sp2 hybridised. Substitution with nitrogen (isoelectronic with sp2 carbon) does not result in a loss of melatonergic activity.
Figure 20 | Swapping N1 and C3 is tolerated
Although the introduction of more polar groups is probably detrimental to pharmacokinetics it may be beneficial from the perspective of total synthesis.
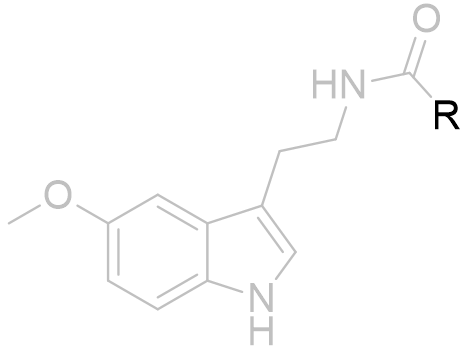
Acetyl group
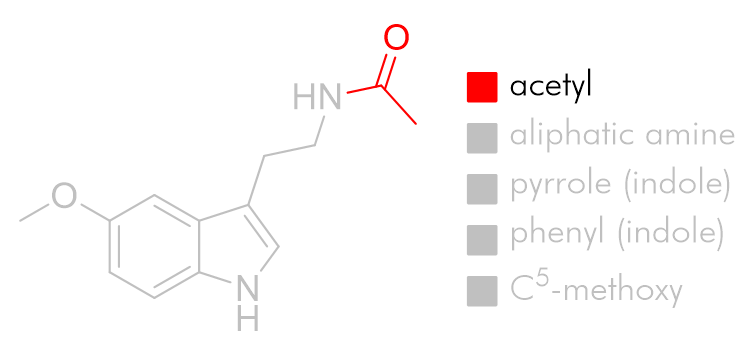
To explore the lipophilic acetyl binding pocket we can draw inferences from the structure activity relationship of acetyl homology to potency. Similar trends are reported for a variety of melatonin analogues and it is generally true that the optimal acetyl chain length for agonists is the butanoyl amide.
 |
Substituent | pEC50 /nM | |
| R=Me |
|
10.00 ± 0.09 | |
| Et | 10.21 ± 0.12 | ||
| nPr | 10.24 ± 0.08 | ||
| Bu | 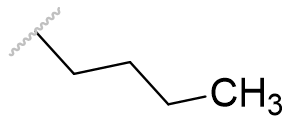 |
8.16 ± 0.20 |
Table 1 | Melatonin acetyl group homologation
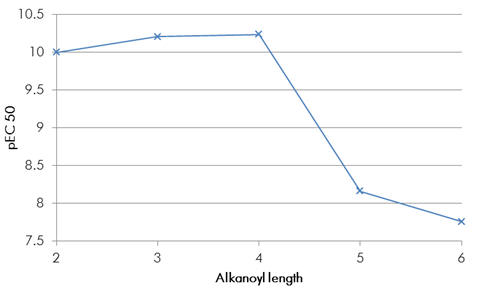
Chart 2 | Melatonin acetyl homologues: butanoyl amide is the optimal
To result in potency gains of this magnitude it is likely the pocket is relatively dense to enable significant gains in induced attractive forces. If the binding site was open to the solvent then it could accommodate even larger groups but also there would be no significant changes to binding or potency. If it is a pocket which binds the acetyl group, then this is deeper than the acetyl group of melatonin would suggest. The cyclobutyl derivative is an antagonist. This comparatively bulky group would not fit the proposed dense binding pocket and so the amide would be prevented from coordination.
 |
R=cBu |  |
All antagonists |
| cPr |  |
||
| Adm | 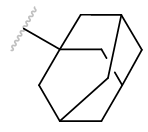 |
Table 2 | Agents with bulky acetyl groups
We can postulate that amide binding has a key role in melatonergic activity. This offers explanation to the observation that acetyl homology does not effect the potency of antagonists. It is also the case that for antagonists despite acceptable acetyl geometries, the homology does not directly correlate with potency. Perhaps this is because of an alternative mode of antagonism, resulting in the absence of amide binding.
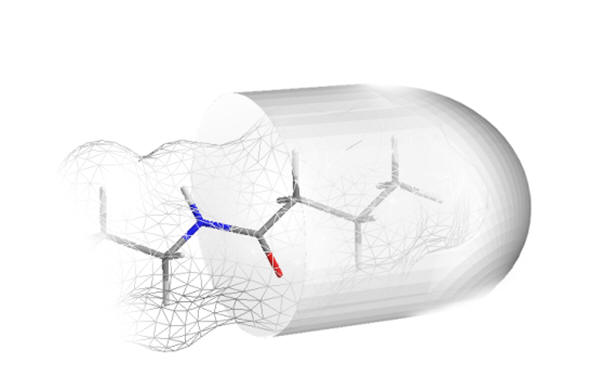 |
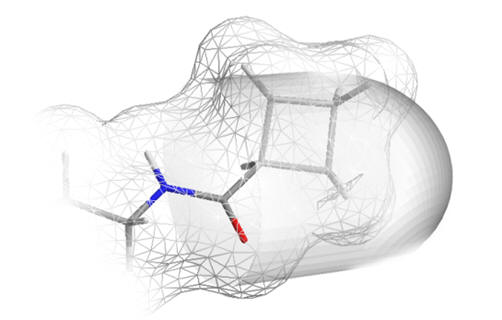 |
| n-chain aliphatics dock neatly | ...whereas bulky groups do not fit |
Figure 21 | The acetyl chain docks into a deep pocket
C5-X substitutions
The C5-methoxy group was once widely regarded to be essential for binding or at least essential for agonist activity. Neither of these postulations are true, although many nor-methoxy analogues are not agonists. However effective agonists lacking the methoxy feature have now been prepared through optimisation of other pharmacophore parts.
Table 3 | Comparison between methoxy and nor-methoxy melatonin
 |
Substituent | EC50 /nM | |
|
|
0.59 ± 0.06 | ||
| 701 ± 93 |
The activity decrease on loss of the methoxy group is partially restored on introduction of 5-bromo but no group has yet been identified which results in an activity rivalling that of the aromatic alkoxy ether parent. This is illustrated by N-methyl melatonin analogues and series of molecular fragments.
Table 4 | The effect of C5-X variation
| Substituent | EC50 /nM | |
 |
 |
0.97 ± 0.20 |
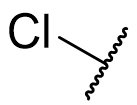 |
2.12 ± 0.01 | |
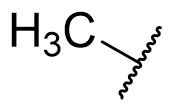 |
16.1 ± 2.7 |
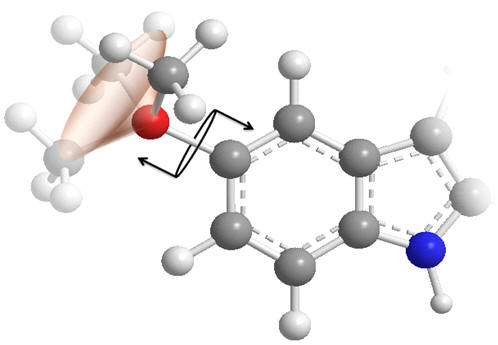
C5-OR conformation
The O-methyl group is not restrained and freely rotates. Despite not being structurally prohibited from attaining any particular dihedral angle (with respect to the aromatic plane), we can make predictions as to which values are more likely to be favoured.
 |
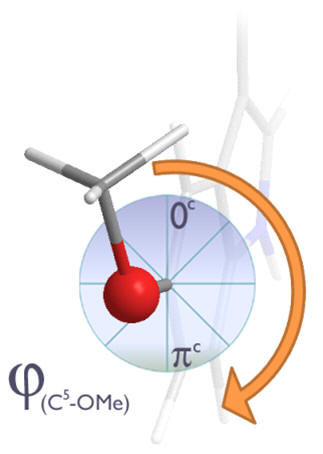 |
Figure 22| Dihedral angles: Un-hindered rotation of the C5-OMe dihedral angle
Concerning the C5-OMe torsional axis, when this dihedral angle subtends by either 0 or πc, the oxygen frontier orbital can conjugate into the aromatic indole system by adopting sp2 hybridisation - at intermediate rotations the un-hybridised p-orbital would become non-bonding and so raise the system energy. We can expect a slight preference for dihedral angles of close to 0 or πc.
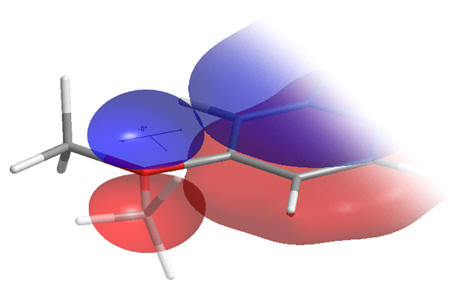 |
 |
| φ = 0, π | φ = ½π, ¾π |
Figure 23 | Possible C5-OMe dihedral angles: Conjugated (left), non-bonding (right)
To ascertain which of the two probable conformations is the active one, the alkyl group was homologated and tethered to either the e-face or the f-face forming a dihydrofuro tricycle.
| Ki /nM | |
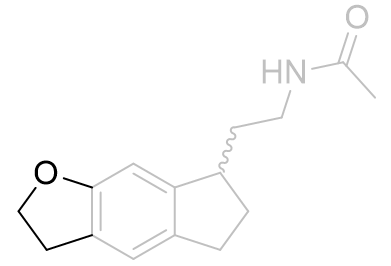 |
483 ± 131 |
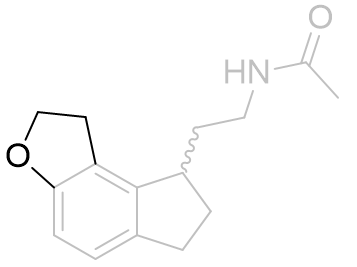 |
0.126 ± 0.035 |
Table 5 | Dihydrofuroindanes: investigation into C5-OR conformation
Restraining the alkyl group to the e-face resulted in a dramatically superior binding affinity compared to the f-face fused analogue. These molecules also obey a similar acetyl homology-activity relationship as do other melatonergic agonists.
| Substituent | EC50 / nM | ||
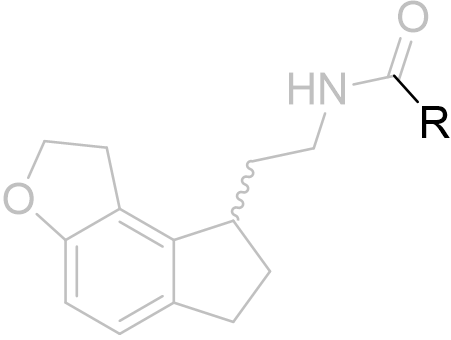 |
R=Me |
|
0.126 ± 0.035 |
| Et |
|
0.0174 ± 0.0041 | |
| Pr |
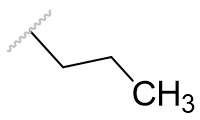 |
0.0214 ± 0.0006 |
Table 6 | Dihydrofuro[e]indane acetyl homology
Lastly the assay of enantiomerically pure isomers demonstrate the optimal configuration is to result in the amide chain subtending below the indole plane
Fragment based studies
To further investigate the roles and significance of N1 and C2 in activity, the elucidation of monocyclic fragment pharmacologies has been pursued.[28] Some small highly effective binding molecules have been identified. Deletion of N1 and C2 does not diminish receptor affinity in a particularly profound way. Thus it can be deduced that the pyrrole moiety plays little role in mediating pharmacodynamic activity, but with a defined but generally tolerant binding pocket can be exploited with the goal of pharmacokinetic tailoring.[26]
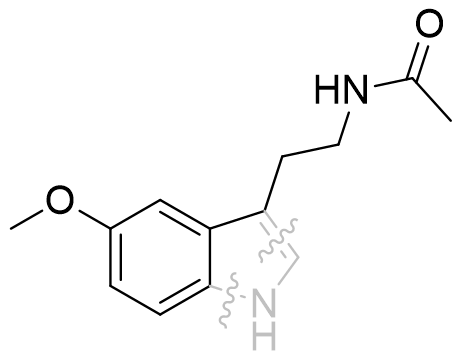
Figure 24 | Melatonin fragment derived by pyrrole disconnection: Ki = 63 ± 4 nM (cf: MLT 0.1nM)
This melatonin fragment also follows the previously observed trend in acetyl homology, with the butanoyl amide binding the strongest (Ki=5.5 ± 1.8) and is surprisingly similar in affinity to melatonin for such a simple molecule.
Table 7 | Molecular fragment investigations lacking the pyrrole moiety
| Ki /nM | |
 |
958 ± 108 |
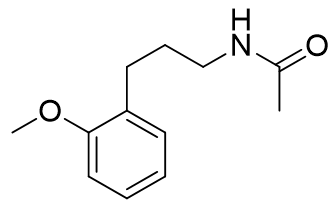 |
1430 ± 310 |
 |
63 ± 4 |
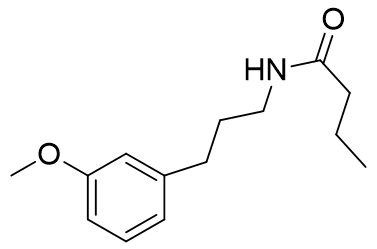 |
5.5 ± 1.8 |
Neither varying aliphatic chain length nor aromatic substitution pattern were well tolerated despite the heteroatom-origin distances remaining comparable. Thus it appears there may be a kink in the receptor around which the molecule must wrap rather than spanning directly or otherwise prevents the simultaneous docking at both receptor sites.
Despite the lack of the pyrrole nucleus, some of these fragment compounds bound strongly. To achieve such efficient binding the receptor must be ordered densely around the ligand.
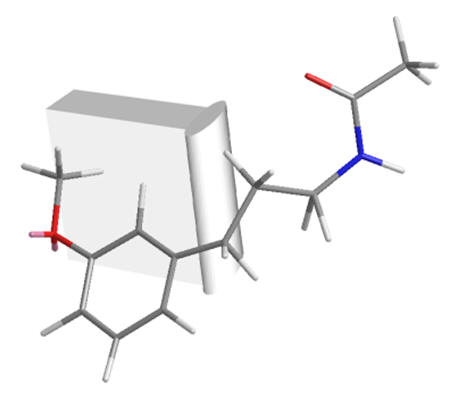 |
 |
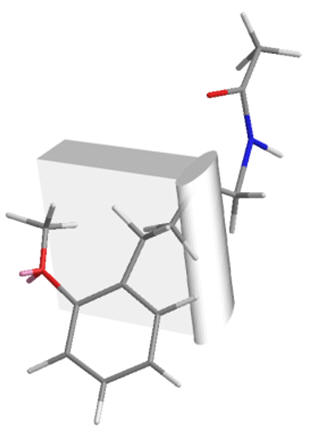 |
| Meta-fragment | Melatonin | Ortho-fragment |
Figure 25 | A bend in the receptor: direct spanning between C5-O and amide is not possible whilst holding the C5-O in the same position
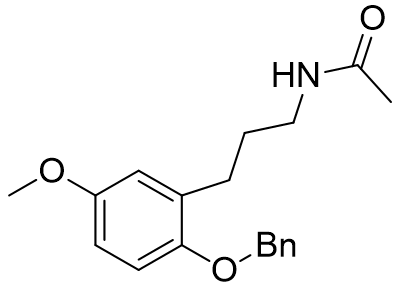
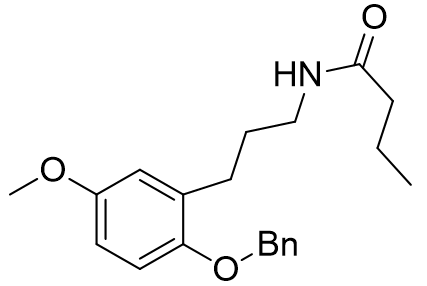
These molecular fragments can also be designed to discriminate between MT1 and MT2 receptors astonishingly well. Longer acetyl groups generally enhance selectivity for MT1.
Table 8 | Honing the selectivity of molecular fragments
| Ki (MT1) /nM | Ki (MT2) /nM | MT1/MT2 | |
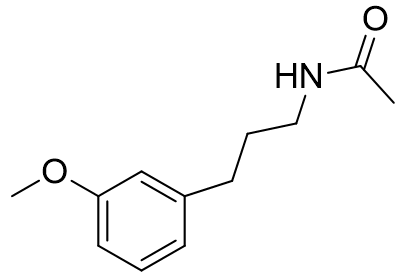 |
23.3 |
0.751 | 31 |
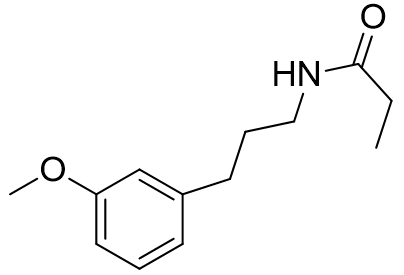 |
85.4 |
7.07 | 12 |
 |
879 |
0.04705 | 1.87 x 104 |
 |
1172 |
0.00103 | 1.14 x 106 |
Incorporation of the benzyloxy substituent simultaneously lowers affinity for MT1 but raises affinity for the MT2. The increased selectivity for the MT1 receptor was particularly marked for the benzyloxy derivatives.[29] This appears to point towards a sterically sensitive MT2 receptor.[15]
C2-X derivatives
Derivatisation of the C2 position is widely tolerated even with quite diverse groups. Most C2 derivatives significantly enhance binding affinity without disrupting pharmacodynamic activity. In fact with the exceptions of benzyl and cyclohexyl, all are more potent agonists than melatonin. Iodomelatonin is an example of a potent agonist (picomolar binding affinity) which is frequently employed as a radioligand for pharmacological characterisation of other agents.
Table 9 | C2 substituents: with the exception of benzyl, all substituents enhanced potency.[30]
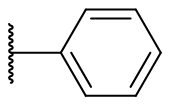
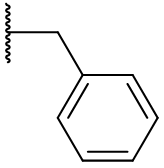
| Substituent | EC50 /nM | Radius/ Å | Volume / Å3 | ||
 |
|
1.1 | 1.1 | 5.8 | |
|
|
0.021 |
 |
2.05 | 36.0 | |
|
|
0.058 | 1.9 | 28.7 | ||
 |
0.057 |
 |
2.6 | 73 | |
|
|
0.43 | 1.5 | 15.6 | ||
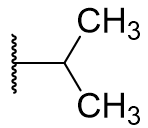 |
0.43 | 1.75 | 22.5 | ||
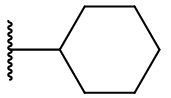 |
5.3 | 3.75 | 179 | ||
 |
antagonist | 3.95 | 258 | ||
All of these substituents are larger than hydrogen and there is a general trend for enhanced dynamics with increasing substituent volume.
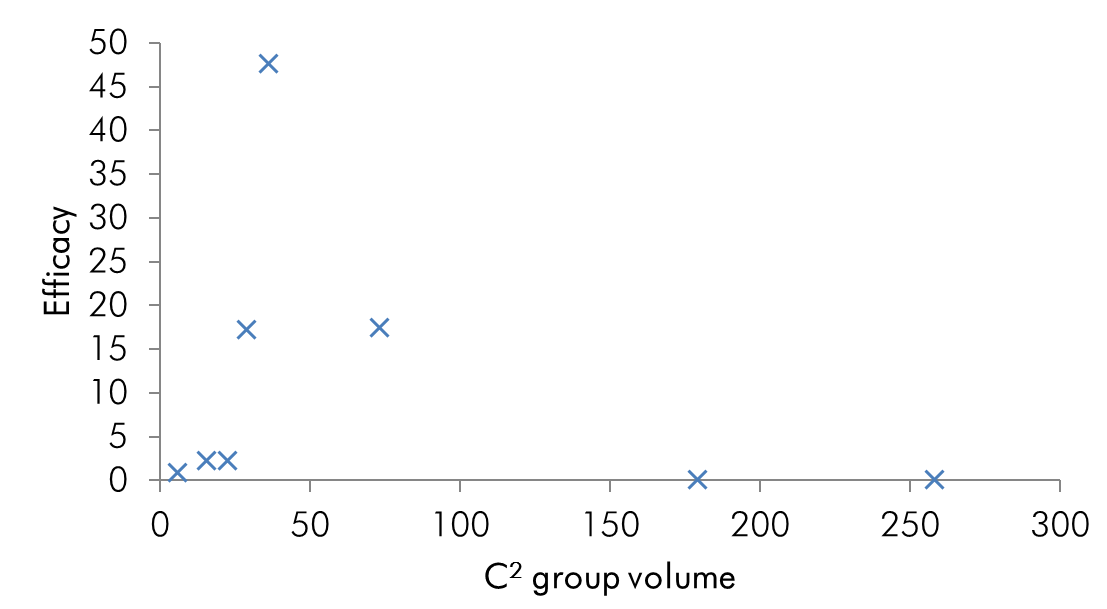
Chart 3 | Potency as a function of C2 substituent volume: the turning point occurs at 36Å indicating the optimal pocket occupancy
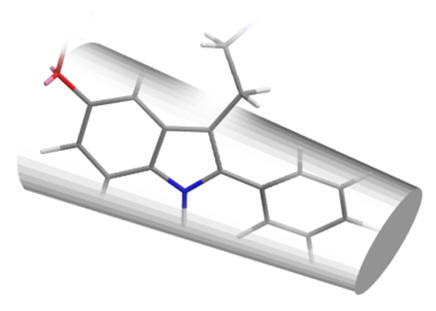
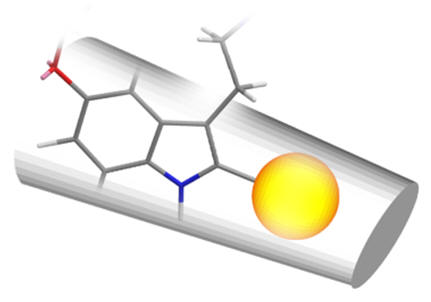
The efficacy of all C2 substituted agonist derivatives implies a spacious C2 environment. Rather than merely tolerating the substituent, the significant increases in activity suggests the accommodation of the group in a closely associating sub-site which may be continuous with the pyrrole/indole binding site. The C2-benzyl derivative is interesting in that the pharmacodynamic activity is reversed: these compounds are antagonists.
 |
 |
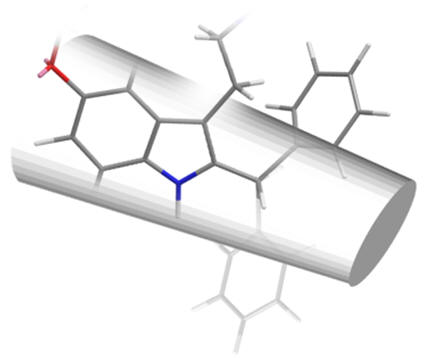 |
| 2-phenyl | 2-iodo | 2-benzyl |
Figure 26 | Schematic of C2 binding pocket: no rotamer of the benzyl compound fits the pocket but those with substituents which extend along the benzo-pyrrole axis do so deeper into the pocket with enhanced dynamics
The loss of activity can perhaps be attributed to a poor insertion into the pocket - the methylene spacer requires approximately tetrahedral bond angles which prohibit linear alignment. This implies that the C2 binding cavity, whilst large, is constrained to a defined volume.
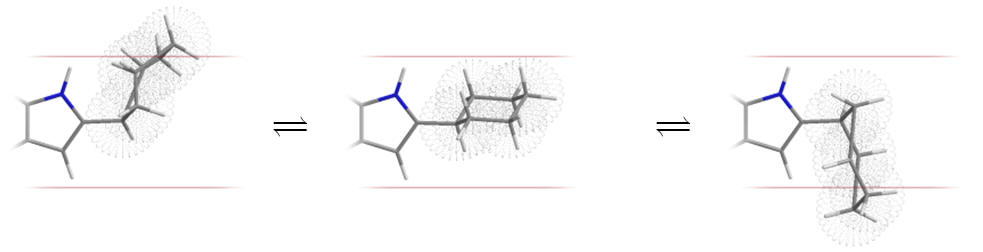
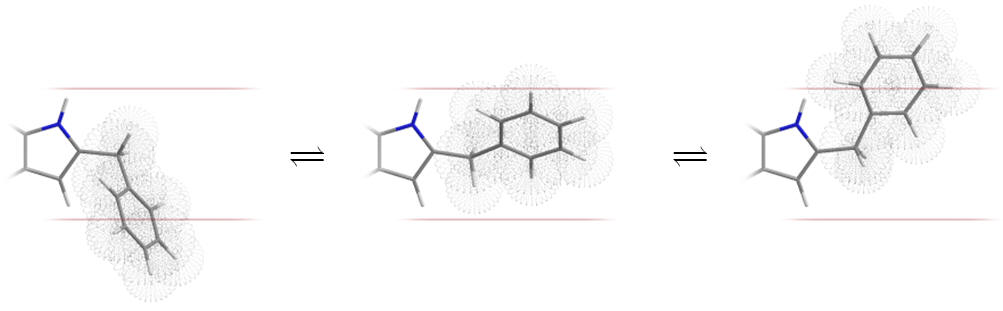
Table 10 | Approximate C2 substituent volume and conformational freedom: because the cyclohexyl and benzyl derivatives are very flexible they can deviate readily from the complementary geometry of the sub-site (as indicated with the boundaries below)
| Substituent | Approximate molecular surface |
|
|
 |
 |
 |
 |
 |
 |
 |
It is possible that an entropic effect hinders the binding of the cyclohexyl and benzyl derivatives. Luzindole is an antagonist based on C2-benzyl nor-methoxy melatonin.[26]
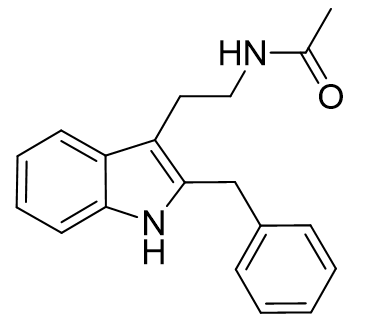
Figure 27 | Luzindole: a potent antagonist which has been instrumental in MLT research
The toleration of the phenyl substituent is a useful feature. It makes the molecule more lipophilic, more potent and enhances binding; subsequently 2-phenylmelatonin has been subject to many structure activity investigations.
2-phenyl melatonin sub-series
Exploiting the enhanced affinity of C2 phenyl derivatisation and the antagonistic dynamic behaviour of cyclobutanoyl amides, N-CBCPT is a powerful antagonist.[26]
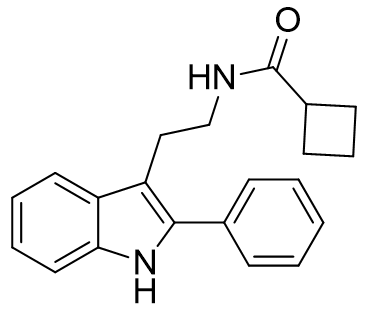
Figure 28 | N-CBCPT
A similar relationship between potency and acetyl homology is observed, comparable to that of the nor-phenyl parent.
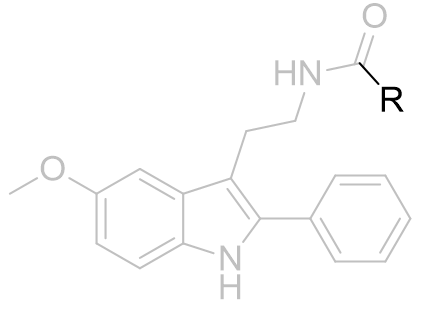
Table 11 | Acetyl group homologation and congeners of 2-phenylmelatonin [31]
| Substituent | pEC50 / nM | Ki /nM | ||
 |
R=Me |
 |
8 | 0.0596 ± 0.01 |
| TFM |
 |
8 | 0.0466 ± 0.0066 | |
| Et |
|
8 | 0.0558 ± 0.012 | |
| cPr |
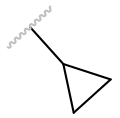 |
Antagonist | 0.0190 ± 0.003 | |
| cBu |
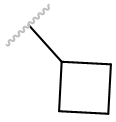 |
Antagonist | 0.3047 ± 0.066 | |
| cPent |
 |
Antagonist | 32.8 ± 7.8 | |
| cHex |
 |
Antagonist | 216 ± 31 |
Face-A fused polycyclic 2-phenylmelatonin derivatives
The N1 and C2 positions locate towards the MLT face-A contact region. The depth of this site has been investigated by phenyl ortho-fusion to N1 affording face-A fused tetracyclic derivatives. Extending the pharmacophore by fusing a benzocyclopentyl bicycle yields an agonist of modest potency but is otherwise unremarkable. Expanding the aliphatic linker to a cyclohexyl decreases the potency, perhaps suggesting the N1 site is shallow or non-continuous with the C2 site. Further expanding the ring to cycloheptyl eliminates all agonist dynamics and is instead an antagonist albeit a low potency one.
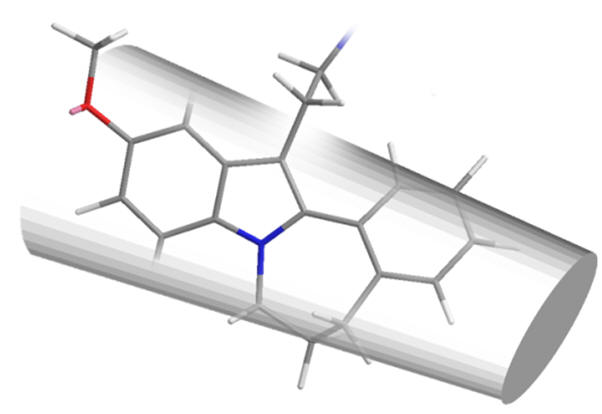
Figure 29 | Large face-A 2-phenyl ortho-fused tetracycles are not well accomodated
These observations are consistent with a sterically demanding face-A binding site. The increasing cyclic interior angle extends the benzo cycle beyond the C2 binding pocket boundaries and this may explain the lack of activity.
Table 12 | Face-A 2-phenyl ortho-fused polycycles
| EC50 /nM | |
 |
2.29 ± 1.0 |
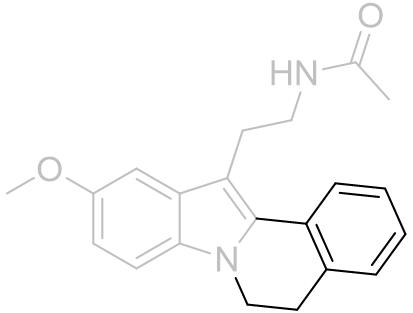 |
12.6 ± 2.12 |
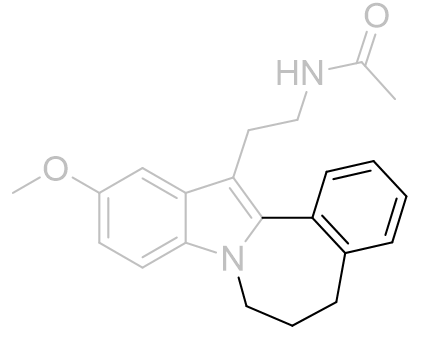 |
antagonist |
The linker restricts rotation of the C2-phenyl bond and this could be interpreted as the C2 site being restricted to particular rotations. However the relatively similar volumes of the spherically symmetric iodo group compared to phenyl does not support this assumption. Instead the face-A site is shallow or otherwise unaccomodating - from the cyclopentyl to the cyclohexyl the linker cycle bulges increasingly against the face-A until binding is impossible.
Aliphatic derivatisation
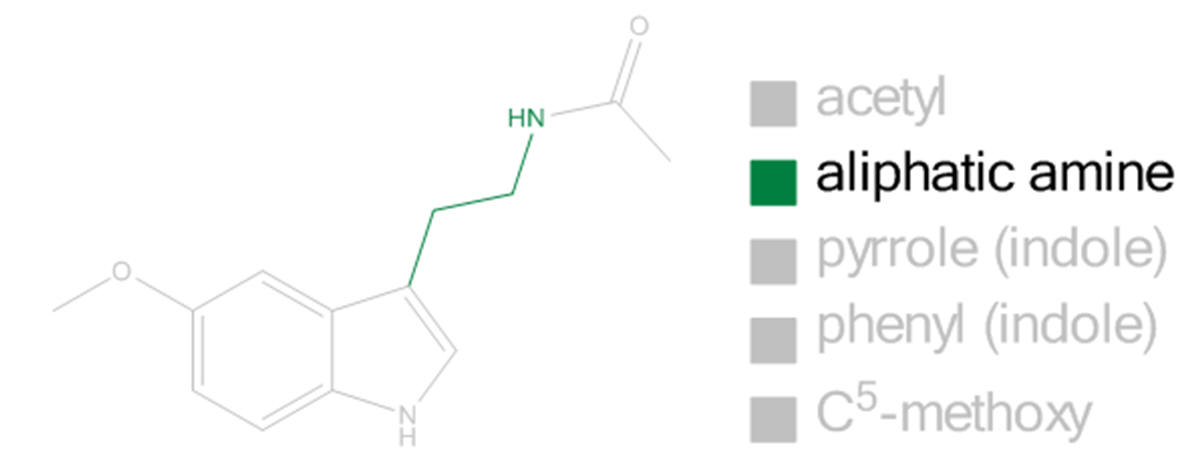
To explore the aliphatic binding groove, inferences are drawn from the studies of aliphatic amine chain derivatisation. The intolerance to variations in steric bulk can be inferred from studies of β-alkyl homologues.
Table 13 | N-methyl melatonin mono-alkyl aliphatic chain derivatisation
| Substituent | EC50 /nM | |
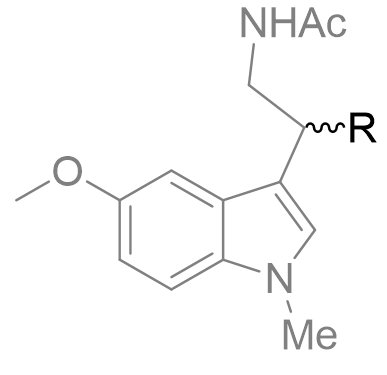 |
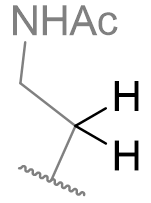 |
0.59 ± 0.06 |
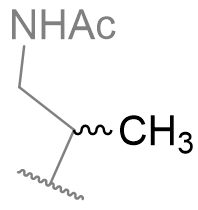 |
1.72 ± 0.08 |
Despite being less potent than the parent, the mono-methyl derivative is interesting because it offers opportunities to investigate the chiral sense of the binding pocket. [33]
Table 14 | β-Monomethyl aliphatic chain derivatives: investigating the site asymmetric sense
| EC50 /nM | EC50 /nM | ||
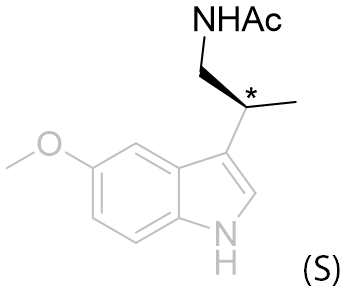 |
10.8 ± 0.08 |
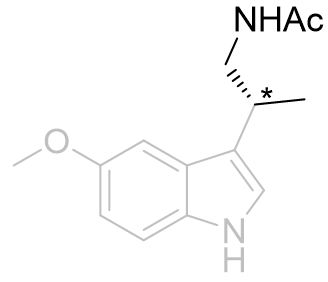 |
0.98 ± 0.01 |
This series suggests the aliphatic region is contoured with a chiral sense and it is observed that the (R) isomer is the better accommodated. To avoid steric clash the amide substituent will probably extend away from the indole. Of the β-substituents the hydrogen atom is the least destabilised when positioned glancing the aromatic ring (syn or gauche). Thus the amide group projects either above, below or in-plane with the ring.
 |
 |
| (S) - amide above the plane | (R) - amide below the plane |
Figure 30 | Newman diagrams: illustration of different amide projections with chiral sense
Beyond this assumption dihedral angles cannot be reliably estimated and further studies of conformationally restrained congeners is necessary, infra.
| Variable | Range | Favoured |
| `phi_((C^3-C^beta))` | `0^c<phi<2pi` | `1/2pi<phi<3/2pi` |
To explore the extent of the aliphatic pocket and to determine what is tolerated, dialkyl derivatives are now considered.
Table 15 | N-methyl melatonin aliphatic chain derivatisation
| Substituent | EC50 /nM | |
 |
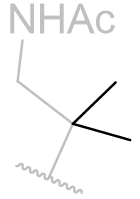 |
6.31 ± 0.94 |
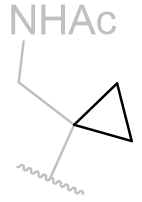 |
10.5 ± 3.0 | |
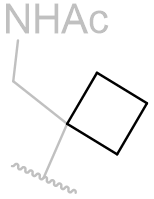 |
1010 ± 210 | |
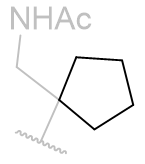 |
antagonist |
In this series of N-methyl melatonin derivatives it is observed that increasing the bulk of the aliphatic chain is to the detriment of activity.
Fused face-B tricyclic derivatives
The positioning of the aliphatic group and the face-B C2 pocket can be jointly probed by tethering the substituent back onto the C2 position forming a fused indolo-tricycle.[26] Also very important is the opportunity these asymmetric molecules present allowing the absolute configuration to be investigated. Changing this face-B fused ring size has significant effects on agent dynamics, but all rings are tolerated.
The efficacy of the cyclohexyl derivative is significantly superior to the others, those both bigger and smaller and is comparable to that of melatonin. This implies that the cyclohexyl derivative has the atoms in locations common to melatonin and that the angles the other rings project the amide group to be intolerable.
Table 16 | Indolo-b-face fused tricycles: aliphatic chain movement restriction
| Ring size | EC50 /nM | Ki | Reference | ||
 |
n=2 |
 |
423 | 161 ± 20 | 5 |
| 3 |
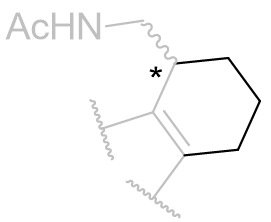 |
0.7 | 0.97 ± 0.2 | 2 | |
| 4 |
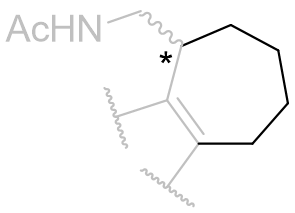 |
258 | 24 ± 3.5 | 5 |
With this C3 angle constrained, the NH-origin distance can be varied specifically by relocating the amide moiety around the ring.[26] At this point it is uncertain whether the amide group subtends below or above the indole plane, but the optimal aliphatic homology (ethylamido) is identical to that of melatonin.
Table 17 | Cyclohexyl[b.3.2]melatonin SAR: aliphatic displacement restriction
| EC50 /nM | Ki | |
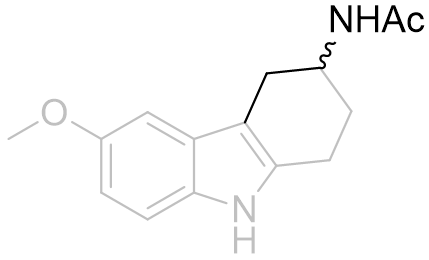 |
32 | 219 ± 50 |
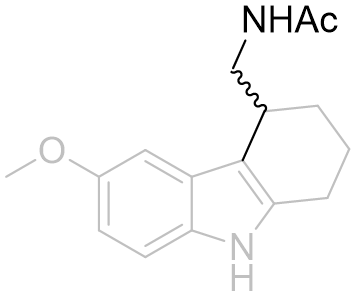 |
0.7 | 0.97 ± 0.2 |
Cyclization from the aliphatic α-position restricts the amide group to distant locations relative to molecular origin whereas β-position derivatisation restricts the amide to more proximal locations. Certainly it appears to be important for the amide to be free to position into tighter conformations as is evidenced by the 500% increase in potency of the β-derivative. This supports the inference that the amide hooks round a receptor kink - for the latter compound of this series this folding is self-evident (see Figure 25).
As racemates the optimal absolute configurations of the aliphatic side-chains are not ascertainable, however one of the stereoisomers of the later must be more potent than either of the stereoisomers of the former. Below are illustrated the possible isomers for comparison: note the degree of backfolding in the compound (D) below.
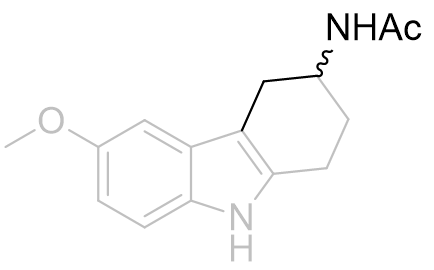 |
 A |
 B |
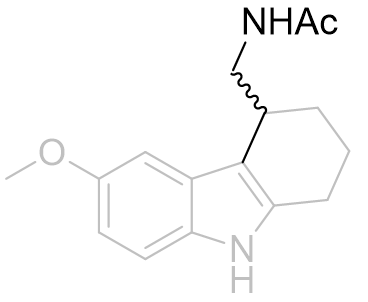 |
 C |
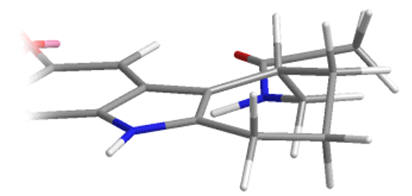 D |
Figure 31 | Cyclohexyl[b.3.2]melatonin: possible aliphatic position and configurations
The chiral sense of this position is very important. Fusion into a tricycle restrains many of the dihedral variables and the absolute configuration thus hints toward the optimal values. The assay of enantiopure stereoisomers demonstrate the optimal dihedral relationship has the amide chain subtending below the indole plane (R-isomer 130 fold more potent).[26] This is in corroboration with the observation of the superiority of the (R)-β-methyl melatonin. On alkylating the indole nitrogen a decrease in activity is observed as is with melatonin.
Table 18 | Enantiopure cyclohexyl[b.3.2]melatonin: investigation into amide chiral sense
| EC50 /nM | Ki | |
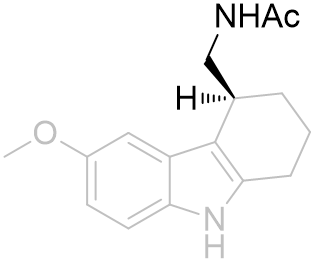 |
48.4 | 219 ± 50 |
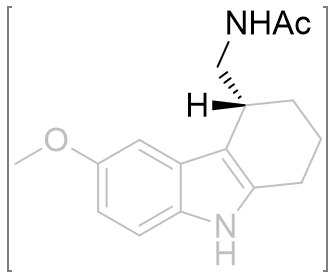 |
0.372 | 0.97 ± 0.2 |
The position of the amide in this pair of compounds is contained to only one variable angle. As the latter is already a particularly potent agonist, if the structure could be restrained further, more of in-effective conformations can be eliminated.
Face-C,D perifused tricyclic derivatives
The position of the amide group can be further clarified by peri-fusion across the C and D faces to form a tricycle.
Table 19 | Perifused-C,D-tricycles
| IC50 /nM | Ki(MT1) | Ki(MT2) | MT1/MT2 | |
 |
9.9 ± 1.2 | 2.7 | 12.15 | 4.5 |
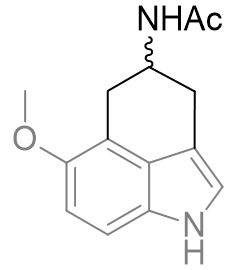 |
1200 ± 80 | 330 | p. agonist | 550 |
There is a very large difference between these binding affinities. From this series we can infer that the amide binding site lays to the side of the pyrrole rather than above the indole. This is supported by molecular overlays which take into account the degree of free rotation and we can visually inspect them to ascertain common geometries.
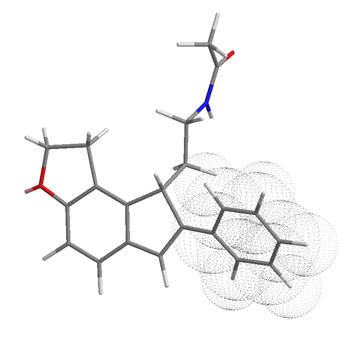
 |
 |
 |
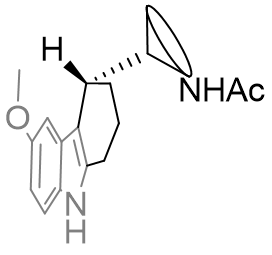 |
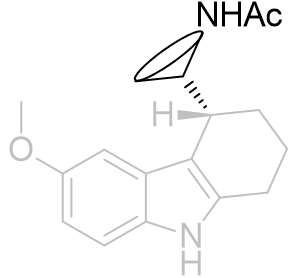
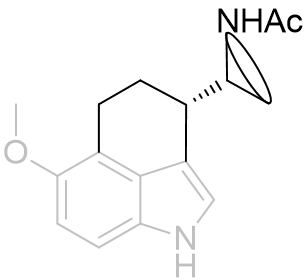
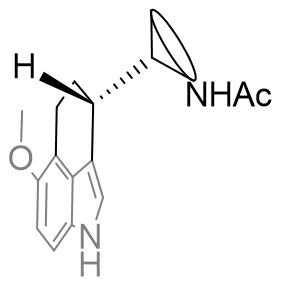
Figure 32 | Comparison between perifused and B-face fused tricycles
If both A and B are potent agonists and both are conformationally restricted then it is likely that their dynamic activity is derived from a common structural or geometric feature. However, features also common with antagonists cannot, if at all, solely be responsible for activity. Following this reasoning the amide substituent in both of these compounds must orient itself into the active conformation which is commonly attainable between both perifused and B-face fused tricycles. This suggests the amide group is orthogonal to the indole plane and subtends below rather than above.
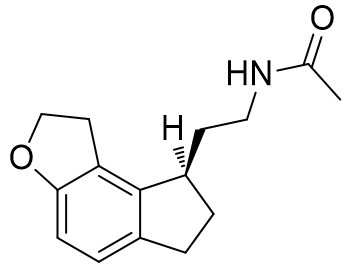
The absolute configuration of the ethylamido side chain can be set to the above but rather than through the introduction of external cycles by saturation at the prochiral C3 position.[27]
Table 20 | Enantiopure dihydrofuro[e]indanes: optimisation of the amide chiral sense
| EC50 /nM | Ki | |
 |
48.4 | 219 ± 50 |
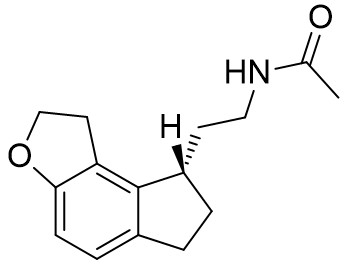 |
0.372 | 0.97 ± 0.2 |
Indeed this series follows the same trend: when the ethylamido chain is held below the indole ring a significantly more potent and strongly binding agent is afforded