The current state of melatonin receptor exploration and exploitation: recent and topical advances
 |
The Design of Melatonergic AgonistsPart 2The Chemistry of Melatoninby Phil |
Contents
- Structure, Definitions and Nomenclature
- Features of the melatonergic pharmacophore
- Conformational analysis
- Opportunities for pharmacophore development
Structure, Definitions and Nomenclature

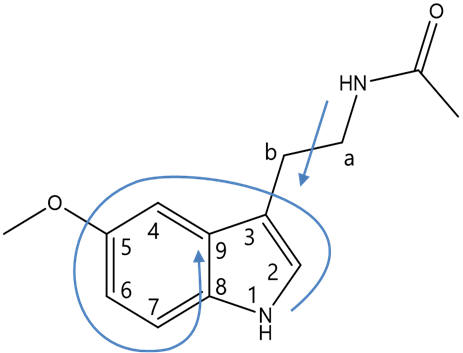
Figure 7 | Structure of melatonin and definitions: face and atom designations
As per the IUPAC convention on systematic nomenclature, the atoms of melatonin are numbered reflecting tryptamine freebase as the root. Hence the indole is attached to the β-carbon of the primary aliphatic chain with the amine group functionalising the α-position. The indole bicycle is then indexed with the highest priority annular atom being the pyrrole like nitrogen (N1) and the atoms numbered sequentially first around the smallest ring (pyrrole) then around the secondary ring (phenyl), skipping the bridgeheads until last. The faces of the molecule are designated alphabetically in Greek, similarly first around the highest priority ring and then round the lower priority ring, again skipping the bridgeheads. Melatonin can be systematically referred to as N-acetyl-5-methoxy tryptamine, but also as a derivative of serotonin can be accurately referred to as N-acetyl-O-methyl serotonin.
 |
 |
| Atom designations | Face designations |
Figure 8 | Structures of melatonin: indicating atom numbers (left) and face designations
Current melatonergic agonists compete with the endogenous ligand for binding and so the agent pharmacophores are best designed to be complementary to the receptor. The intelligent design of bioactive agents de novo therefore requires corroboration with a working model of the target receptor.
From first principles the features of melatonin which could potentially facilitate molecular interactions can be recognized. These will not be necessarily optimal but they provide the precedence and direction for exploratory investigation. A process of ligand variation then allows the systematic elucidation of the roles of each component of the ligand. The major binding modes and can be identified, optimised and exploited. Structure activity relationships help ascertain features that are key to agent dynamics (ie., trigger groups) and also the optimal structural homologies and conformations.
Tolerated changes can be used to modify the chemical properties affecting the pharmacokinetic profile. These inferences are used to advance the working hypothesis which enables the design of new agents for experiments to further explore the receptor, take the hypothesis forwards and produce lead compounds.
Features of the melatonergic pharmacophore
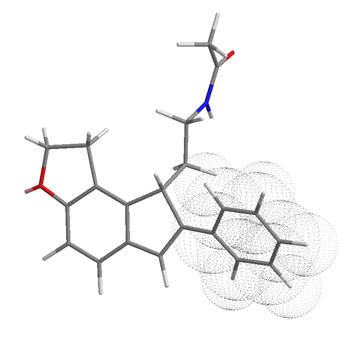
Melatonin is a largely lipophilic molecule. The central indole nucleus is aromatic and this has to capacity to associate to the receptor in a π-π stacking fashion. The indole is certainly best accommodated by other planar aromatic systems (Trp, Phe, Tyr etc.) as alternatively hybridised groups (ie., Ala, Glu, Asp, Val etc.) are thermodynamically restricted from presenting an equivalently stable surface. A bicyclic motif is nearly ubiquitous amongst melatonergic molecules.
The aliphatic terminal of the acetyl group and similarly the C5O-alkyl terminal could potentially extend into lipophilic pockets. The primary aliphatic chain is likely to span an aliphatic region locating the amide into its sub-site.
In addition to the van der Waals and π-π stacking interactions, melatonin is capable of hydrogen-bonding. This is likely to be a major mode of receptor interaction but as a neuroactive tryptophan derivative melatonin is unusual. Rather than a mono-amine base like dopamine, ephedrine or serotonin it is an amide. Acetylation of the terminal α-amine of 5-methoxyserotonin radically changes the hydrogen bonding behaviour. The lone-pair of the aliphatic nitrogen atom is involved in mesomerism and this disables its H-bond acceptor ability. Likewise the capping of serotonins 5-hydroxy group disables the ability to act as an H-bond donor. Relative to the mono-amines, this is a reversal of H-bond character. Potential H-bond acceptors include the C5-O and the amide-carbonyl oxygen; donors of H-bonding include the indole N1-H and the amide Nα-H. Reflecting these differences to the mono-amines, melatonergic molecules are mostly N-acetylated, 5-O-alkylate derivatives. This maintains drug-receptor binding interactions consistent with those of melatonin.

Figure 10 | H-bonding interactions: differences caused by O-alkylation and N-acetylation
Conformational analysis
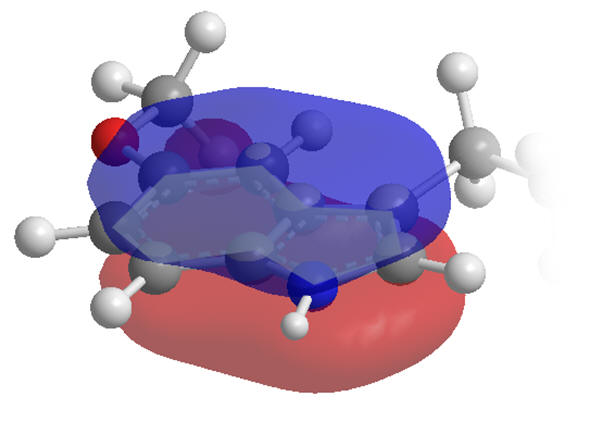
The indole moiety is restrained to a planar conformation due to delocalisation across the aromatic system. Internal rotation of the primary amide ethanoyl group is also restricted but by mesomerism between amide and acetimidic forms as the nitrogen lone-pairs delocalise into the carbonyl group. Melatonin is not however an entirely rigid molecule.
 |
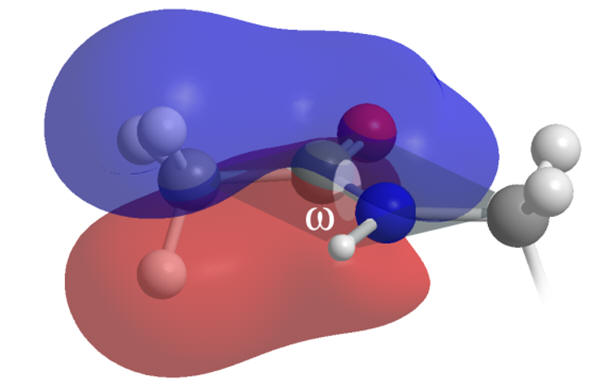 |
Figure 11 | Planar and rigid regions of melatonin: aromatic indole (left) & amide (right)
Rotation about the carbanoyl-amine axis (ω) is unfavourable as it proceeds via a de-conjugated transition state and so the amide group is flat.
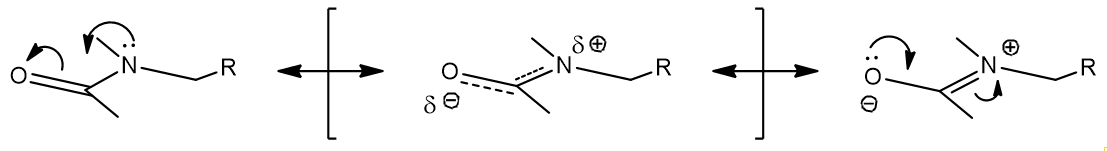
Figure 12 | Amide resonance structures: mesomerism between amide and acetimidic acid
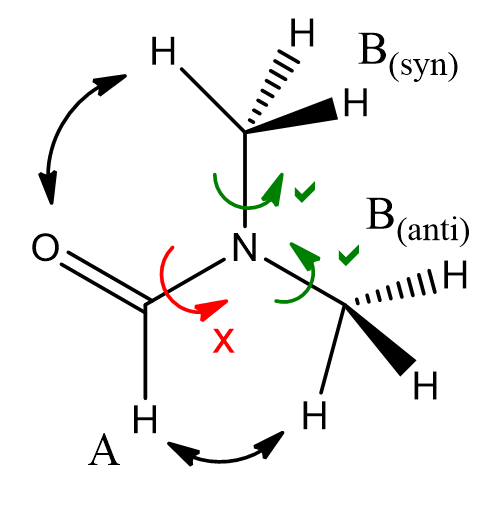
Restricted rotation of amides is a fundamental effect in protein chemistry and is evidenced by low-temperature/thermal programme NMR experiments.
 |
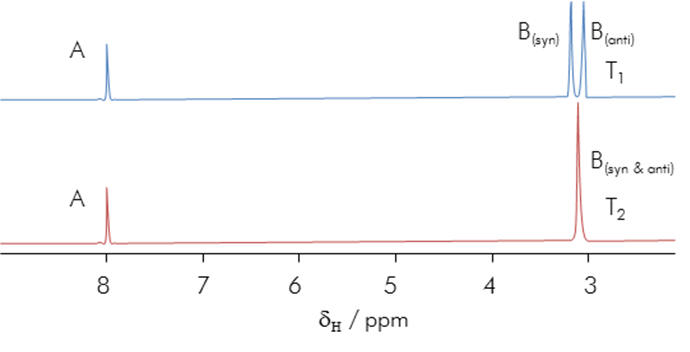 |
Figure 13 | Amide rotational constraint: Thermally prohibitive
rotation breaks the degeneracy of N substituent environments in DMF.
Distinct N-substituent signals are observed in the NMR spectrum at low
temperature but these coalesce at elevated temperatures (T2 > T1)
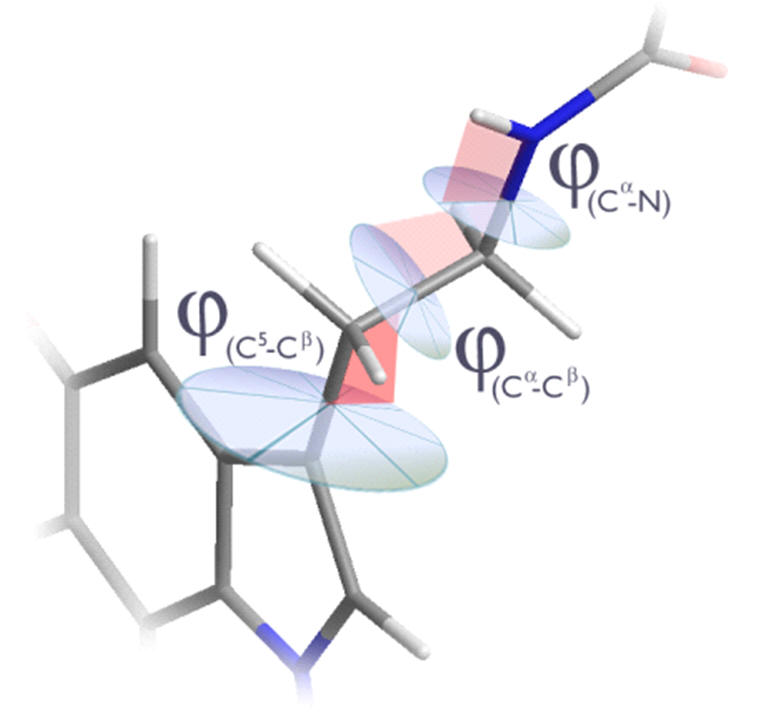
The nature of the aliphatic conformation is far less easily rationalised. There are three independent dihedral angles, none of which are constrained and so there is an entropic propensity to deviate from any particular conformation.
 |
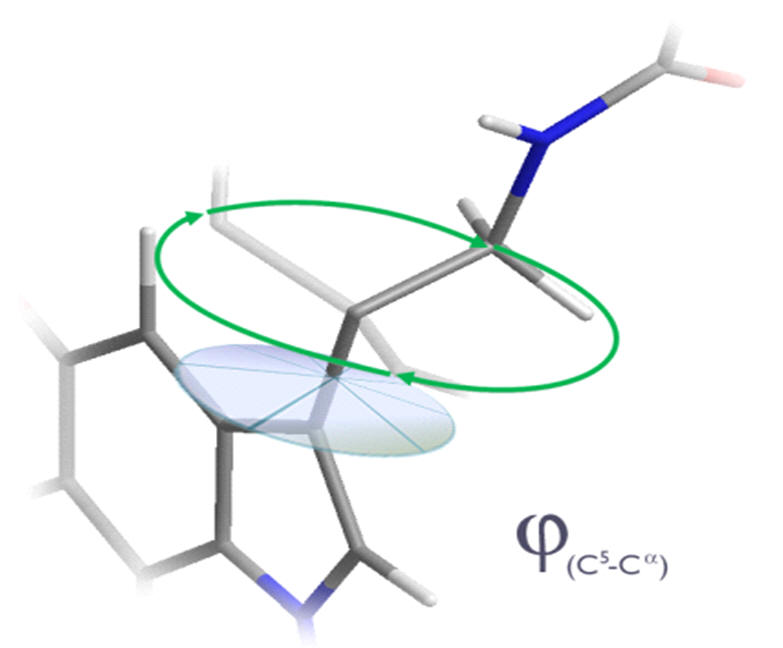 |
Figure 14 | Aliphatic conformation: Independant dihedral angles and free rotation
Opportunities for pharmacophore development
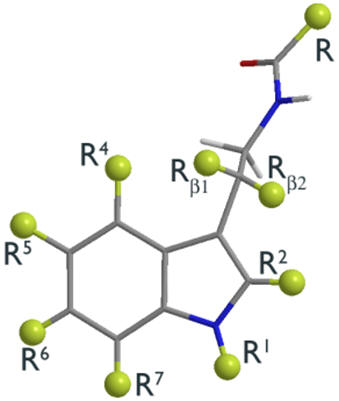
Melatonin as a structural platform offers many opportunities for pharmacophore variation. Catenae and annuli homologation, bio-isosteric substitution, derivatisation and the configuration of absolute conformation are all tailorable facets.
 |
 |
 |
| Peripheral substituents | Annular atoms | Di-hedral angles |
Figure 15 | The scope of structural variation
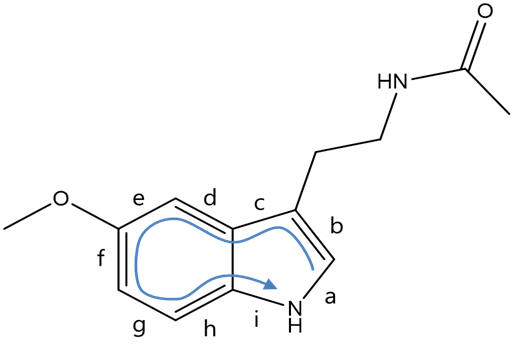
A comprehensive analysis of all possible variables is beyond the scope of this review. Those included are limited to the annular indole atoms of the pyrrole subunit, the indole substituent groups (R1 to R7), aliphatic substituents (Rβ1 and Rβ2) and the acetyl chain, R and their relative positions as determined by their dihedral angles (φs and ω) and absolute configurations.
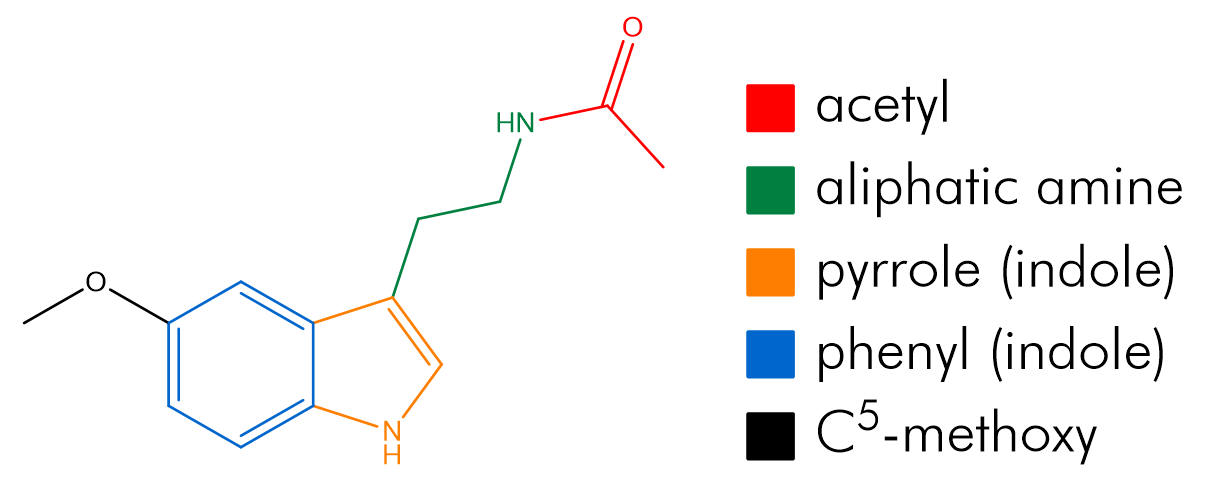
Figure 16 | Regions of Melatonin
Notwithstanding melatonins achiral structure, asymmetric centres are incredibly important to investigate. Opportunities for the introduction of chirality must be seized upon because they provide information on the asymmetry of the receptor and active ligand conformation.
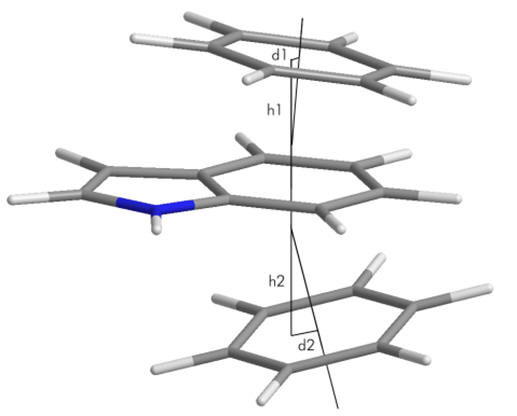
A universal parameterisation for SARs studies is the absolute atomic displacement of particular atoms from the face of fusion. In all polycyclic structures this point serves as a common origin that can be identified and thus allows the comparison of diverse structures.
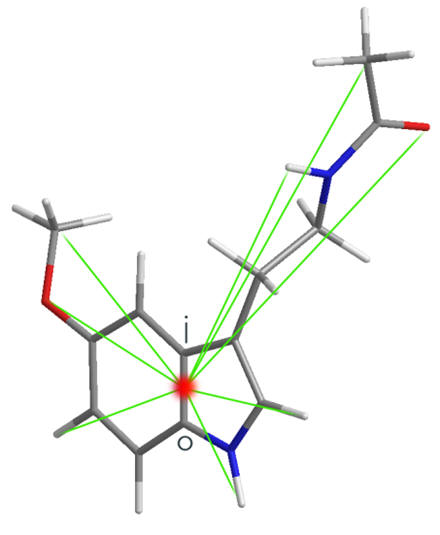
Figure 17 | Structural parameterisation: atom-origin displacements